BioSQL — это общая схема базы данных, предназначенная главным образом для хранения последовательностей и связанных с ними данных для всего ядра СУБД. Он спроектирован таким образом, что в нем хранятся данные из всех популярных баз данных по биоинформатике, таких как GenBank, Swissport и т. Д. Он также может использоваться для хранения собственных данных.
В настоящее время BioSQL предоставляет специальную схему для следующих баз данных:
- MySQL (biosqldb-mysql.sql)
- PostgreSQL (biosqldb-pg.sql)
- Oracle (biosqldb-ora / *. Sql)
- SQLite (biosqldb-sqlite.sql)
Он также обеспечивает минимальную поддержку баз данных HSQLDB и Derby на основе Java.
BioPython предоставляет очень простые, легкие и расширенные возможности ORM для работы с базой данных на основе BioSQL. BioPython предоставляет модуль BioSQL для выполнения следующих функций:
- Создать / удалить базу данных BioSQL
- Подключиться к базе данных BioSQL
- Проанализируйте базу данных последовательностей, такую как GenBank, Swisport, результат BLAST, результат Entrez и т. Д., И непосредственно загрузите ее в базу данных BioSQL.
- Получить данные последовательности из базы данных BioSQL
- Получить данные таксономии из NCBI BLAST и сохранить их в базе данных BioSQL
- Выполнить любой SQL-запрос к базе данных BioSQL
Обзор схемы базы данных BioSQL
Прежде чем углубляться в BioSQL, давайте разберемся с основами схемы BioSQL. Схема BioSQL предоставляет более 25 таблиц для хранения данных последовательности, функции последовательности, категории последовательности / онтологии и информации о таксономии. Вот некоторые из важных таблиц:
- biodatabase
- bioentry
- biosequence
- seqfeature
- таксон
- taxon_name
- Антология
- срок
- dxref
Создание базы данных BioSQL
В этом разделе мы создадим образец базы данных BioSQL, biosql, используя схему, предоставленную командой BioSQL. Мы будем работать с базой данных SQLite, так как это действительно легко начать и не имеет сложной настройки.
Здесь мы создадим базу данных BioSQL на основе SQLite, используя следующие шаги.
Шаг 1 — Загрузите движок базы данных SQLite и установите его.
Шаг 2 — Загрузите проект BioSQL с URL GitHub. https://github.com/biosql/biosql
Шаг 3 — Откройте консоль и создайте каталог с помощью mkdir и войдите в него.
cd /path/to/your/biopython/sample mkdir sqlite-biosql cd sqlite-biosql
Шаг 4 — Запустите приведенную ниже команду, чтобы создать новую базу данных SQLite.
> sqlite3.exe mybiosql.db SQLite version 3.25.2 2018-09-25 19:08:10 Enter ".help" for usage hints. sqlite>
Шаг 5 — Скопируйте файл biosqldb-sqlite.sql из проекта BioSQL (/ sql / biosqldb-sqlite.sql`) и сохраните его в текущем каталоге.
Шаг 6 — Запустите приведенную ниже команду, чтобы создать все таблицы.
sqlite> .read biosqldb-sqlite.sql
Теперь все таблицы созданы в нашей новой базе данных.
Шаг 7 — Запустите команду ниже, чтобы увидеть все новые таблицы в нашей базе данных.
sqlite> .headers on sqlite> .mode column sqlite> .separator ROW "\n" sqlite> SELECT name FROM sqlite_master WHERE type = 'table'; biodatabase taxon taxon_name ontology term term_synonym term_dbxref term_relationship term_relationship_term term_path bioentry bioentry_relationship bioentry_path biosequence dbxref dbxref_qualifier_value bioentry_dbxref reference bioentry_reference comment bioentry_qualifier_value seqfeature seqfeature_relationship seqfeature_path seqfeature_qualifier_value seqfeature_dbxref location location_qualifier_value sqlite>
Первые три команды являются командами конфигурации для настройки SQLite для отображения результата в отформатированном виде.
Шаг 8 — Скопируйте образец файла GenBank, ls_orchid.gbk, предоставленный командой BioPython https://raw.githubusercontent.com/biopython/biopython/master/Doc/examples/ls_orchid.gbk, в текущий каталог и сохраните его как orchid.gbk ,
Шаг 9 — Создайте скрипт python load_orchid.py, используя приведенный ниже код, и выполните его.
from Bio import SeqIO from BioSQL import BioSeqDatabase import os server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db") db = server.new_database("orchid") count = db.load(SeqIO.parse("orchid.gbk", "gb"), True) server.commit() server.close()
Приведенный выше код анализирует запись в файле и преобразует ее в объекты Python и вставляет ее в базу данных BioSQL. Мы проанализируем код в следующем разделе.
Наконец, мы создали новую базу данных BioSQL и загрузили в нее несколько примеров данных. Мы обсудим важные таблицы в следующей главе.
Простая схема ER
Таблица базы данных находится в верхней части иерархии, и ее основное назначение — организовать набор данных последовательности в одну группу / виртуальную базу данных. Каждая запись в базе данных biod относится к отдельной базе данных и не смешивается с другой базой данных. Все связанные таблицы в базе данных BioSQL имеют ссылки на запись в базе данных.
Таблица bioentry содержит все детали о последовательности, кроме данных о последовательности. Данные о последовательности конкретной био-записи будут сохранены в таблице биопоследовательности .
taxon и taxon_name являются деталями таксономии, и каждая запись ссылается на эту таблицу для указания информации о таксонах.
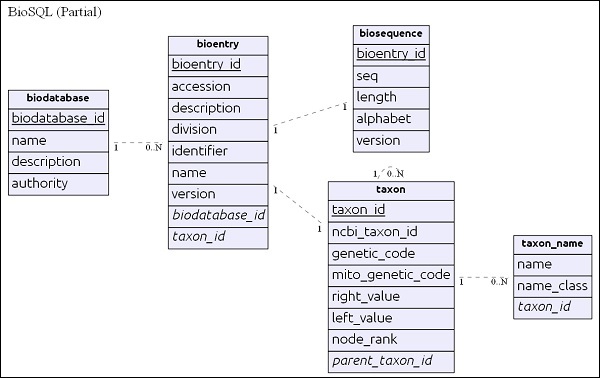
После понимания схемы, давайте посмотрим на некоторые запросы в следующем разделе.
Запросы BioSQL
Давайте углубимся в некоторые SQL-запросы, чтобы лучше понять, как организованы данные и как таблицы связаны друг с другом. Прежде чем продолжить, давайте откроем базу данных с помощью приведенной ниже команды и установим некоторые команды форматирования:
> sqlite3 orchid.db SQLite version 3.25.2 2018-09-25 19:08:10 Enter ".help" for usage hints. sqlite> .header on sqlite> .mode columns
.header и .mode — параметры форматирования для лучшей визуализации данных . Вы также можете использовать любой редактор SQLite для запуска запроса.
Перечислите базу данных виртуальных последовательностей, доступную в системе, как указано ниже —
select * from biodatabase; *** Result *** sqlite> .width 15 15 15 15 sqlite> select * from biodatabase; biodatabase_id name authority description --------------- --------------- --------------- --------------- 1 orchid sqlite>
Здесь у нас есть только одна база данных, орхидея .
Перечислите записи (топ 3), доступные в базе данных orchid с приведенным ниже кодом
select
be.*,
bd.name
from
bioentry be
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid' Limit 1,
3;
*** Result ***
sqlite> .width 15 15 10 10 10 10 10 50 10 10
sqlite> select be.*, bd.name from bioentry be inner join biodatabase bd on
bd.biodatabase_id = be.biodatabase_id where bd.name = 'orchid' Limit 1,3;
bioentry_id biodatabase_id taxon_id name accession identifier division description version name
--------------- --------------- ---------- ---------- ---------- ---------- ----------
---------- ---------- ----------- ---------- --------- ---------- ----------
2 1 19 Z78532 Z78532 2765657 PLN
C.californicum 5.8S rRNA gene and ITS1 and ITS2 DN 1
orchid
3 1 20 Z78531 Z78531 2765656 PLN
C.fasciculatum 5.8S rRNA gene and ITS1 and ITS2 DN 1
orchid
4 1 21 Z78530 Z78530 2765655 PLN
C.margaritaceum 5.8S rRNA gene and ITS1 and ITS2 D 1
orchid
sqlite>
Перечислите подробности последовательности, связанные с записью (номер доступа — Z78530, название — ген C. Fasciculatum 5.8S рРНК и ДНК ITS1 и ITS2) с указанным кодом —
select
substr(cast(bs.seq as varchar), 0, 10) || '...' as seq,
bs.length,
be.accession,
be.description,
bd.name
from
biosequence bs
inner join
bioentry be
on be.bioentry_id = bs.bioentry_id
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid'
and be.accession = 'Z78532';
*** Result ***
sqlite> .width 15 5 10 50 10
sqlite> select substr(cast(bs.seq as varchar), 0, 10) || '...' as seq,
bs.length, be.accession, be.description, bd.name from biosequence bs inner
join bioentry be on be.bioentry_id = bs.bioentry_id inner join biodatabase bd
on bd.biodatabase_id = be.biodatabase_id where bd.name = 'orchid' and
be.accession = 'Z78532';
seq length accession description name
------------ ---------- ---------- ------------ ------------ ---------- ---------- -----------------
CGTAACAAG... 753 Z78532 C.californicum 5.8S rRNA gene and ITS1 and ITS2 DNA orchid
sqlite>
Получите полную последовательность, связанную с записью (инвентарный номер — Z78530, название — ген C. Fasciculatum 5.8S рРНК и ДНК ITS1 и ITS2), используя приведенный ниже код —
select
bs.seq
from
biosequence bs
inner join
bioentry be
on be.bioentry_id = bs.bioentry_id
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid'
and be.accession = 'Z78532';
*** Result ***
sqlite> .width 1000
sqlite> select bs.seq from biosequence bs inner join bioentry be on
be.bioentry_id = bs.bioentry_id inner join biodatabase bd on bd.biodatabase_id =
be.biodatabase_id where bd.name = 'orchid' and be.accession = 'Z78532';
seq
----------------------------------------------------------------------------------------
----------------------------
CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGTTGAGACAACAGAATATATGATCGAGTGAATCT
GGAGGACCTGTGGTAACTCAGCTCGTCGTGGCACTGCTTTTGTCGTGACCCTGCTTTGTTGTTGGGCCTCC
TCAAGAGCTTTCATGGCAGGTTTGAACTTTAGTACGGTGCAGTTTGCGCCAAGTCATATAAAGCATCACTGATGAATGACATTATTGT
CAGAAAAAATCAGAGGGGCAGTATGCTACTGAGCATGCCAGTGAATTTTTATGACTCTCGCAACGGATATCTTGGCTC
TAACATCGATGAAGAACGCAG
sqlite>
Список таксонов, связанных с био базой орхидей
select distinct tn.name from biodatabase d inner join bioentry e on e.biodatabase_id = d.biodatabase_id inner join taxon t on t.taxon_id = e.taxon_id inner join taxon_name tn on tn.taxon_id = t.taxon_id where d.name = 'orchid' limit 10; *** Result *** sqlite> select distinct tn.name from biodatabase d inner join bioentry e on e.biodatabase_id = d.biodatabase_id inner join taxon t on t.taxon_id = e.taxon_id inner join taxon_name tn on tn.taxon_id = t.taxon_id where d.name = 'orchid' limit 10; name ------------------------------ Cypripedium irapeanum Cypripedium californicum Cypripedium fasciculatum Cypripedium margaritaceum Cypripedium lichiangense Cypripedium yatabeanum Cypripedium guttatum Cypripedium acaule pink lady's slipper Cypripedium formosanum sqlite>
Загрузка данных в базу данных BioSQL
Давайте узнаем, как загрузить данные последовательности в базу данных BioSQL в этой главе. У нас уже есть код для загрузки данных в базу данных в предыдущем разделе, и код выглядит следующим образом:
from Bio import SeqIO
from BioSQL import BioSeqDatabase
import os
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
DBSCHEMA = "biosqldb-sqlite.sql"
SQL_FILE = os.path.join(os.getcwd(), DBSCHEMA)
server.load_database_sql(SQL_FILE)
server.commit()
db = server.new_database("orchid")
count = db.load(SeqIO.parse("orchid.gbk", "gb"), True) server.commit()
server.close()
Мы будем более подробно рассматривать каждую строку кода и его цель —
Линия 1 — загружает модуль SeqIO.
Строка 2 — загружает модуль BioSeqDatabase. Этот модуль предоставляет все функциональные возможности для взаимодействия с базой данных BioSQL.
Линия 3 — Загрузка модуля ОС.
Строка 5 — open_database открывает указанную базу данных (db) с настроенным драйвером (драйвер) и возвращает дескриптор базы данных BioSQL (сервер). Biopython поддерживает базы данных sqlite, mysql, postgresql и oracle.
Строка 6-10 — метод load_database_sql загружает sql из внешнего файла и выполняет его. Метод commit фиксирует транзакцию. Мы можем пропустить этот шаг, потому что мы уже создали базу данных со схемой.
Строка 12 — методы new_database создают новую виртуальную базу данных orchid и возвращают дескриптор db для выполнения команды в базе данных orchid.
Строка 13 — метод load загружает записи последовательности (итерируемый SeqRecord) в базу данных orchid. SqlIO.parse анализирует базу данных GenBank и возвращает все последовательности в ней как повторяемый SeqRecord. Второй параметр (True) в методе загрузки указывает ему на получение сведений о таксономии данных последовательности с веб-сайта взрыва NCBI, если они еще не доступны в системе.
Строка 14 — commit фиксирует транзакцию.
Строка 15 — close закрывает соединение с базой данных и уничтожает дескриптор сервера.
Получить данные последовательности
Давайте выберем последовательность с идентификатором 2765658 из базы данных орхидей, как показано ниже:
from BioSQL import BioSeqDatabase
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
db = server["orchid"]
seq_record = db.lookup(gi = 2765658)
print(seq_record.id, seq_record.description[:50] + "...")
print("Sequence length %i," % len(seq_record.seq))
Здесь server [«orchid»] возвращает дескриптор для извлечения данных из виртуальной базы данных orchid. Метод lookup предоставляет возможность выбирать последовательности на основе критериев, и мы выбрали последовательность с идентификатором 2765658. lookup возвращает информацию о последовательности в виде SeqRecordobject. Поскольку мы уже знаем, как работать с SeqRecord`, легко получить данные из него.
Удалить базу данных
Удалить базу данных так же просто, как вызвать метод remove_database с правильным именем базы данных, а затем зафиксировать его, как указано ниже —