Биопайтон — Введение
Biopython — это самый большой и самый популярный пакет биоинформатики для Python. Он содержит ряд различных подмодулей для общих задач биоинформатики. Он разработан Чепменом и Чангом, в основном написан на Python. Он также содержит C-код для оптимизации сложной вычислительной части программного обеспечения. Он работает на Windows, Linux, Mac OS X и т. Д.
По сути, Biopython — это набор модулей Python, которые предоставляют функции для работы с последовательностями ДНК, РНК и белковых последовательностей, такими как обратное комплементация цепочки ДНК, поиск мотивов в белковых последовательностях и т. Д. Он предоставляет множество синтаксических анализаторов для чтения всех основных генетических баз данных. такие как GenBank, SwissPort, FASTA и т. д., а также оболочки / интерфейсы для запуска других популярных программ / инструментов биоинформатики, таких как NCBI BLASTN, Entrez и т. д., в среде Python. У него есть родственные проекты, такие как BioPerl, BioJava и BioRuby.
Характеристики
Biopython является портативным, понятным и имеет простой в освоении синтаксис. Некоторые из существенных особенностей перечислены ниже —
-
Интерпретируемый, интерактивный и объектно-ориентированный.
-
Поддерживает FASTA, PDB, GenBank, Blast, SCOP, PubMed / Medline, ExPASy.
-
Возможность работать с форматами последовательности.
-
Инструменты для управления белковыми структурами.
-
BioSQL — стандартный набор таблиц SQL для хранения последовательностей, а также функций и аннотаций.
-
Доступ к онлайн-сервисам и базе данных, включая сервисы NCBI (Blast, Entrez, PubMed) и сервисы ExPASY (SwissProt, Prosite).
-
Доступ к местным услугам, включая Blast, Clustalw, EMBOSS.
Интерпретируемый, интерактивный и объектно-ориентированный.
Поддерживает FASTA, PDB, GenBank, Blast, SCOP, PubMed / Medline, ExPASy.
Возможность работать с форматами последовательности.
Инструменты для управления белковыми структурами.
BioSQL — стандартный набор таблиц SQL для хранения последовательностей, а также функций и аннотаций.
Доступ к онлайн-сервисам и базе данных, включая сервисы NCBI (Blast, Entrez, PubMed) и сервисы ExPASY (SwissProt, Prosite).
Доступ к местным услугам, включая Blast, Clustalw, EMBOSS.
цели
Цель Biopython — предоставить простой, стандартный и расширенный доступ к биоинформатике через язык Python. Конкретные цели Биопиона перечислены ниже —
-
Обеспечение стандартизированного доступа к ресурсам биоинформатики.
-
Высококачественные, многоразовые модули и скрипты.
-
Быстрое манипулирование массивом, которое можно использовать в коде кластера, PDB, NaiveBayes и модели Маркова.
-
Анализ геномных данных.
Обеспечение стандартизированного доступа к ресурсам биоинформатики.
Высококачественные, многоразовые модули и скрипты.
Быстрое манипулирование массивом, которое можно использовать в коде кластера, PDB, NaiveBayes и модели Маркова.
Анализ геномных данных.
преимущества
Biopython требует очень мало кода и имеет следующие преимущества:
-
Предоставляет тип данных микрочипа, используемый в кластеризации.
-
Читает и пишет файлы типа Tree-View.
-
Поддерживает структурные данные, используемые для анализа, представления и анализа PDB.
-
Поддерживает данные журнала, используемые в приложениях Medline.
-
Поддерживает базу данных BioSQL, которая широко используется в качестве стандартной базы данных среди всех проектов в области биоинформатики.
-
Поддерживает разработку синтаксического анализатора, предоставляя модули для анализа файла биоинформатики в объект записи определенного формата или универсальный класс последовательности плюс функции.
-
Четкая документация, основанная на стиле поваренной книги.
Предоставляет тип данных микрочипа, используемый в кластеризации.
Читает и пишет файлы типа Tree-View.
Поддерживает структурные данные, используемые для анализа, представления и анализа PDB.
Поддерживает данные журнала, используемые в приложениях Medline.
Поддерживает базу данных BioSQL, которая широко используется в качестве стандартной базы данных среди всех проектов в области биоинформатики.
Поддерживает разработку синтаксического анализатора, предоставляя модули для анализа файла биоинформатики в объект записи определенного формата или универсальный класс последовательности плюс функции.
Четкая документация, основанная на стиле поваренной книги.
Пример тематического исследования
Давайте проверим некоторые варианты использования (популяционная генетика, структура РНК и т. Д.) И попытаемся понять, как Biopython играет важную роль в этой области —
Популяционная генетика
Популяционная генетика — это изучение генетической изменчивости в популяции, которая включает изучение и моделирование изменений частот генов и аллелей в популяциях в пространстве и времени.
Biopython предоставляет модуль Bio.PopGen для популяционной генетики. Этот модуль содержит все необходимые функции для сбора информации о классической популяционной генетике.
Структура РНК
Три основные биологические макромолекулы, которые необходимы для нашей жизни, это ДНК, РНК и белок. Белки являются рабочими лошадками клетки и играют важную роль в качестве ферментов. ДНК (дезоксирибонуклеиновая кислота) считается «светокопией» клетки. Он несет всю генетическую информацию, необходимую для роста клетки, потребления питательных веществ и размножения. РНК (рибонуклеиновая кислота) действует как «фотокопия ДНК» в клетке.
Biopython предоставляет объекты Bio.Sequence, которые представляют нуклеотиды, строительные блоки ДНК и РНК.
Биопайтон — Установка
В этом разделе объясняется, как установить Biopython на ваш компьютер. Он очень прост в установке и займет не более пяти минут.
Шаг 1 — Проверка установки Python
Biopython предназначен для работы с Python 2.5 или более поздними версиями. Таким образом, обязательно, чтобы Python был установлен первым. Запустите приведенную ниже команду в командной строке —
> python --version
Это определено ниже —
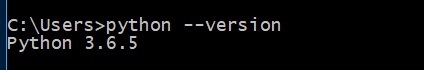
Он показывает версию Python, если установлен правильно. В противном случае загрузите последнюю версию python, установите его и затем снова введите команду.
Шаг 2 — Установка Biopython с помощью pip
Biopython легко установить с помощью pip из командной строки на всех платформах. Введите следующую команду —
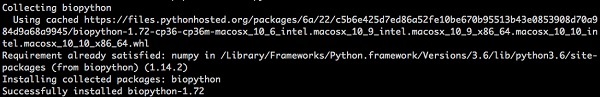
> pip install biopython
Следующий ответ будет виден на вашем экране —
Для обновления старой версии Biopython —
> pip install biopython –-upgrade
Следующий ответ будет виден на вашем экране —
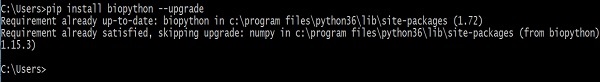
После выполнения этой команды более старые версии Biopython и NumPy (от этого зависит Biopython) будут удалены перед установкой последних версий.
Шаг 3 — Проверка установки Biopython
Теперь вы успешно установили Biopython на свой компьютер. Чтобы убедиться, что Biopython установлен правильно, введите следующую команду на консоли python:
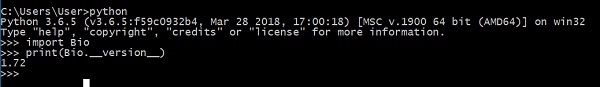
Показывает версию Biopython.
Альтернативный способ — установка Biopython с использованием Source
Чтобы установить Biopython с использованием исходного кода, следуйте приведенным ниже инструкциям —
Загрузите последний выпуск Biopython по следующей ссылке — https://biopython.org/wiki/Download
На данный момент последней версией является biopython-1.72 .
Загрузите файл и распакуйте сжатый архивный файл, перейдите в папку с исходным кодом и введите следующую команду —
> python setup.py build
Это создаст Biopython из исходного кода, как указано ниже —
Теперь протестируйте код с помощью следующей команды —
> python setup.py test
Наконец, установите с помощью следующей команды —
> python setup.py install
Biopython — Создание простого приложения
Давайте создадим простое приложение Biopython для анализа файла биоинформатики и распечатки содержимого. Это поможет нам понять общую концепцию биопиона и то, как он помогает в области биоинформатики.
Шаг 1 — Сначала создайте пример файла последовательности «example.fasta» и поместите в него содержимое ниже.
>sp|P25730|FMS1_ECOLI CS1 fimbrial subunit A precursor (CS1 pilin) MKLKKTIGAMALATLFATMGASAVEKTISVTASVDPTVDLLQSDGSALPNSVALTYSPAV NNFEAHTINTVVHTNDSDKGVVVKLSADPVLSNVLNPTLQIPVSVNFAGKPLSTTGITID SNDLNFASSGVNKVSSTQKLSIHADATRVTGGALTAGQYQGLVSIILTKSTTTTTTTKGT >sp|P15488|FMS3_ECOLI CS3 fimbrial subunit A precursor (CS3 pilin) MLKIKYLLIGLSLSAMSSYSLAAAGPTLTKELALNVLSPAALDATWAPQDNLTLSNTGVS NTLVGVLTLSNTSIDTVSIASTNVSDTSKNGTVTFAHETNNSASFATTISTDNANITLDK NAGNTIVKTTNGSQLPTNLPLKFITTEGNEHLVSGNYRANITITSTIKGGGTKKGTTDKK
Расширение fasta относится к формату файла последовательности. FASTA происходит от программного обеспечения для биоинформатики, FASTA и, следовательно, получает свое имя. Формат FASTA состоит из нескольких последовательностей, расположенных одна за другой, и каждая последовательность будет иметь свой собственный идентификатор, имя, описание и фактические данные последовательности.
Шаг 2 — Создайте новый скрипт на Python * simple_example.py «, введите приведенный ниже код и сохраните его.
from Bio.SeqIO import parse from Bio.SeqRecord import SeqRecord from Bio.Seq import Seq file = open("example.fasta") records = parse(file, "fasta") for record in records: print("Id: %s" % record.id) print("Name: %s" % record.name) print("Description: %s" % record.description) print("Annotations: %s" % record.annotations) print("Sequence Data: %s" % record.seq) print("Sequence Alphabet: %s" % record.seq.alphabet)
Давайте немного углубимся в код —
Строка 1 импортирует класс разбора, доступный в модуле Bio.SeqIO. Модуль Bio.SeqIO используется для чтения и записи файла последовательности в другом формате, а класс parse используется для анализа содержимого файла последовательности.
Строка 2 импортирует класс SeqRecord, доступный в модуле Bio.SeqRecord. Этот модуль используется для управления записями последовательности, а класс SeqRecord используется для представления определенной последовательности, доступной в файле последовательности.
* Строка 3 « импортирует класс Seq, доступный в модуле Bio.Seq. Этот модуль используется для манипулирования данными последовательности, а класс Seq используется для представления данных последовательности конкретной записи последовательности, доступной в файле последовательности.
Строка 5 открывает файл «example.fasta» с помощью обычной функции python, open.
Строка 7 анализирует содержимое файла последовательности и возвращает содержимое в виде списка объекта SeqRecord.
Строка 9-15 циклически перебирает записи, используя python for loop, и печатает атрибуты записи последовательности (SqlRecord), такие как id, имя, описание, данные последовательности и т. Д.
Строка 15 печатает тип последовательности, используя класс Alphabet.
Шаг 3 — Откройте командную строку и перейдите в папку, содержащую файл последовательности, «example.fasta» и выполните следующую команду —
> python simple_example.py
Шаг 4 — Python запускает скрипт и печатает все данные последовательности, доступные в файле примера, «example.fasta». Вывод будет похож на следующий контент.
Id: sp|P25730|FMS1_ECOLI Name: sp|P25730|FMS1_ECOLI Decription: sp|P25730|FMS1_ECOLI CS1 fimbrial subunit A precursor (CS1 pilin) Annotations: {} Sequence Data: MKLKKTIGAMALATLFATMGASAVEKTISVTASVDPTVDLLQSDGSALPNSVALTYSPAVNNFEAHTINTVVHTNDSD KGVVVKLSADPVLSNVLNPTLQIPVSVNFAGKPLSTTGITIDSNDLNFASSGVNKVSSTQKLSIHADATRVTGGALTA GQYQGLVSIILTKSTTTTTTTKGT Sequence Alphabet: SingleLetterAlphabet() Id: sp|P15488|FMS3_ECOLI Name: sp|P15488|FMS3_ECOLI Decription: sp|P15488|FMS3_ECOLI CS3 fimbrial subunit A precursor (CS3 pilin) Annotations: {} Sequence Data: MLKIKYLLIGLSLSAMSSYSLAAAGPTLTKELALNVLSPAALDATWAPQDNLTLSNTGVSNTLVGVLTLSNTSIDTVS IASTNVSDTSKNGTVTFAHETNNSASFATTISTDNANITLDKNAGNTIVKTTNGSQLPTNLPLKFITTEGNEHLVSGN YRANITITSTIKGGGTKKGTTDKK Sequence Alphabet: SingleLetterAlphabet()
В этом примере мы видели три класса: parse, SeqRecord и Seq. Эти три класса предоставляют большую часть функциональности, и мы изучим эти классы в следующем разделе.
Биопайтон — последовательность
Последовательность — это серия букв, используемых для представления белка, ДНК или РНК организма. Он представлен классом Seq. Класс Seq определен в модуле Bio.Seq.
Давайте создадим простую последовательность в Biopython, как показано ниже —
>>> from Bio.Seq import Seq >>> seq = Seq("AGCT") >>> seq Seq('AGCT') >>> print(seq) AGCT
Здесь мы создали простую белковую последовательность AGCT, и каждая буква представляет собой ланин, G- лизин, цистеин и т- хреонин.
Каждый объект Seq имеет два важных атрибута:
-
данные — фактическая последовательность строк (AGCT)
-
алфавит — используется для представления типа последовательности. например, последовательность ДНК, последовательность РНК и т. д. По умолчанию она не представляет какую-либо последовательность и носит общий характер.
данные — фактическая последовательность строк (AGCT)
алфавит — используется для представления типа последовательности. например, последовательность ДНК, последовательность РНК и т. д. По умолчанию она не представляет какую-либо последовательность и носит общий характер.
Алфавитный модуль
Объекты Seq содержат атрибут Alphabet для указания типа последовательности, букв и возможных операций. Это определяется в модуле Bio.Alphabet. Алфавит может быть определен как ниже —
>>> from Bio.Seq import Seq >>> myseq = Seq("AGCT") >>> myseq Seq('AGCT') >>> myseq.alphabet Alphabet()
Алфавитный модуль предоставляет ниже классы для представления различных типов последовательностей. Алфавит — базовый класс для всех типов алфавитов.
SingleLetterAlphabet — Общий алфавит с буквами размера один. Это происходит от Алфавита, и все другие типы алфавитов происходят из него.
>>> from Bio.Seq import Seq >>> from Bio.Alphabet import single_letter_alphabet >>> test_seq = Seq('AGTACACTGGT', single_letter_alphabet) >>> test_seq Seq('AGTACACTGGT', SingleLetterAlphabet())
ProteinAlphabet — Общий однобуквенный белковый алфавит.
>>> from Bio.Seq import Seq >>> from Bio.Alphabet import generic_protein >>> test_seq = Seq('AGTACACTGGT', generic_protein) >>> test_seq Seq('AGTACACTGGT', ProteinAlphabet())
NucleotideAlphabet — Общий однобуквенный нуклеотидный алфавит.
>>> from Bio.Seq import Seq >>> from Bio.Alphabet import generic_nucleotide >>> test_seq = Seq('AGTACACTGGT', generic_nucleotide) >>> test_seq Seq('AGTACACTGGT', NucleotideAlphabet())
DNAAlphabet — Общий однобуквенный алфавит ДНК.
>>> from Bio.Seq import Seq >>> from Bio.Alphabet import generic_dna >>> test_seq = Seq('AGTACACTGGT', generic_dna) >>> test_seq Seq('AGTACACTGGT', DNAAlphabet())
RNAAlphabet — Общий однобуквенный алфавит РНК.
>>> from Bio.Seq import Seq >>> from Bio.Alphabet import generic_rna >>> test_seq = Seq('AGTACACTGGT', generic_rna) >>> test_seq Seq('AGTACACTGGT', RNAAlphabet())
Модуль Biopython, Bio.Alphabet.IUPAC предоставляет базовые типы последовательностей, определенные сообществом IUPAC. Он содержит следующие классы —
-
ИЮПАКПротеин (белок) — белок ИЮПАК алфавит из 20 стандартных аминокислот.
-
ExtendedIUPACProtein (extended_protein) — расширенный заглавный алфавит с одним буквой IUPAC, включая X.
-
IUPACAmbiguousDNA (ambiguous_dna) — Прописная IUPAC неоднозначная ДНК.
-
IUPACUnambiguousDNA (unambiguous_dna) — Прописная IUPAC однозначная ДНК (GATC).
-
ExtendedIUPACDNA (extended_dna) — расширенный алфавит ДНК IUPAC.
-
IUPACAmbiguousRNA (ambiguous_rna) — Прописная IUPAC неоднозначная РНК.
-
IUPACUnambiguousRNA (однозначная_рна) — прописная IUPAC однозначная РНК (GAUC).
ИЮПАКПротеин (белок) — белок ИЮПАК алфавит из 20 стандартных аминокислот.
ExtendedIUPACProtein (extended_protein) — расширенный заглавный алфавит с одним буквой IUPAC, включая X.
IUPACAmbiguousDNA (ambiguous_dna) — Прописная IUPAC неоднозначная ДНК.
IUPACUnambiguousDNA (unambiguous_dna) — Прописная IUPAC однозначная ДНК (GATC).
ExtendedIUPACDNA (extended_dna) — расширенный алфавит ДНК IUPAC.
IUPACAmbiguousRNA (ambiguous_rna) — Прописная IUPAC неоднозначная РНК.
IUPACUnambiguousRNA (однозначная_рна) — прописная IUPAC однозначная РНК (GAUC).
Рассмотрим простой пример для класса IUPACProtein, как показано ниже —
>>> from Bio.Alphabet import IUPAC >>> protein_seq = Seq("AGCT", IUPAC.protein) >>> protein_seq Seq('AGCT', IUPACProtein()) >>> protein_seq.alphabet
Кроме того, Biopython предоставляет все данные конфигурации, связанные с биоинформатикой, через модуль Bio.Data. Например, IUPACData.protein_letters имеет возможные буквы алфавита IUPACProtein.
>>> from Bio.Data import IUPACData >>> IUPACData.protein_letters 'ACDEFGHIKLMNPQRSTVWY'
Основные операции
В этом разделе кратко объясняются все основные операции, доступные в классе Seq. Последовательности похожи на строки Python. Мы можем выполнять операции со строками Python, такие как нарезка, подсчет, объединение, поиск, разбиение и разбиение на последовательности.
Используйте приведенные ниже коды для получения различных выходов.
Чтобы получить первое значение в последовательности.
>>> seq_string = Seq("AGCTAGCT") >>> seq_string[0] 'A'
Для печати первых двух значений.
>>> seq_string[0:2] Seq('AG')
Чтобы распечатать все значения.
>>> seq_string[ : ] Seq('AGCTAGCT')
Выполнять операции длины и счета.
>>> len(seq_string) 8 >>> seq_string.count('A') 2
Добавить две последовательности.
>>> from Bio.Alphabet import generic_dna, generic_protein >>> seq1 = Seq("AGCT", generic_dna) >>> seq2 = Seq("TCGA", generic_dna) >>> seq1+seq2 Seq('AGCTTCGA', DNAAlphabet())
Здесь два вышеупомянутых объекта последовательности, seq1, seq2, являются общими последовательностями ДНК, поэтому вы можете добавить их и создать новую последовательность. Вы не можете добавлять последовательности с несовместимыми алфавитами, такими как последовательность белка и последовательность ДНК, как указано ниже —
>>> dna_seq = Seq('AGTACACTGGT', generic_dna) >>> protein_seq = Seq('AGUACACUGGU', generic_protein) >>> dna_seq + protein_seq ..... ..... TypeError: Incompatible alphabets DNAAlphabet() and ProteinAlphabet() >>>
Чтобы добавить две или более последовательности, сначала сохраните его в списке Python, затем извлеките его с помощью цикла for и, наконец, добавьте его вместе, как показано ниже:
>>> from Bio.Alphabet import generic_dna >>> list = [Seq("AGCT",generic_dna),Seq("TCGA",generic_dna),Seq("AAA",generic_dna)] >>> for s in list: ... print(s) ... AGCT TCGA AAA >>> final_seq = Seq(" ",generic_dna) >>> for s in list: ... final_seq = final_seq + s ... >>> final_seq Seq('AGCTTCGAAAA', DNAAlphabet())
В следующем разделе приведены различные коды для получения выходных данных в соответствии с требованиями.
Чтобы изменить регистр последовательности.
>>> from Bio.Alphabet import generic_rna >>> rna = Seq("agct", generic_rna) >>> rna.upper() Seq('AGCT', RNAAlphabet())
Проверить членство в Python и личность оператора.
>>> rna = Seq("agct", generic_rna) >>> 'a' in rna True >>> 'A' in rna False >>> rna1 = Seq("AGCT", generic_dna) >>> rna is rna1 False
Найти одну букву или последовательность букв внутри заданной последовательности.
>>> protein_seq = Seq('AGUACACUGGU', generic_protein) >>> protein_seq.find('G') 1 >>> protein_seq.find('GG') 8
Выполнить операцию расщепления.
>>> protein_seq = Seq('AGUACACUGGU', generic_protein) >>> protein_seq.split('A') [Seq('', ProteinAlphabet()), Seq('GU', ProteinAlphabet()), Seq('C', ProteinAlphabet()), Seq('CUGGU', ProteinAlphabet())]
Для выполнения полосовых операций в последовательности.
>>> strip_seq = Seq(" AGCT ") >>> strip_seq Seq(' AGCT ') >>> strip_seq.strip() Seq('AGCT')
Biopython — Расширенные операции с последовательностями
В этой главе мы обсудим некоторые расширенные функции последовательности, предоставляемые Biopython.
Дополнение и обратное дополнение
Нуклеотидная последовательность может быть обратно дополнена, чтобы получить новую последовательность. Кроме того, дополненная последовательность может быть обратно дополнена, чтобы получить исходную последовательность. Biopython предоставляет два метода для выполнения этой функции — дополнение и reverse_complement . Код для этого приведен ниже —
>>> from Bio.Alphabet import IUPAC >>> nucleotide = Seq('TCGAAGTCAGTC', IUPAC.ambiguous_dna) >>> nucleotide.complement() Seq('AGCTTCAGTCAG', IUPACAmbiguousDNA()) >>>
Здесь метод комплемента () позволяет дополнить последовательность ДНК или РНК. Метод reverse_complement () дополняет и переворачивает результирующую последовательность слева направо. Это показано ниже —
>>> nucleotide.reverse_complement() Seq('GACTGACTTCGA', IUPACAmbiguousDNA())
Biopython использует переменную ambiguous_dna_complement, предоставленную Bio.Data.IUPACData, для выполнения операции дополнения.
>>> from Bio.Data import IUPACData >>> import pprint >>> pprint.pprint(IUPACData.ambiguous_dna_complement) { 'A': 'T', 'B': 'V', 'C': 'G', 'D': 'H', 'G': 'C', 'H': 'D', 'K': 'M', 'M': 'K', 'N': 'N', 'R': 'Y', 'S': 'S', 'T': 'A', 'V': 'B', 'W': 'W', 'X': 'X', 'Y': 'R'} >>>
GC Content
Прогнозируется, что базовый состав геномной ДНК (содержание GC) существенно влияет на функционирование генома и экологию видов. Содержание GC представляет собой количество нуклеотидов GC, деленное на общее количество нуклеотидов.
Чтобы получить содержание нуклеотидов ГХ, импортируйте следующий модуль и выполните следующие шаги:
>>> from Bio.SeqUtils import GC >>> nucleotide = Seq("GACTGACTTCGA",IUPAC.unambiguous_dna) >>> GC(nucleotide) 50.0
транскрипция
Транскрипция — это процесс изменения последовательности ДНК в последовательность РНК. Фактический процесс биологической транскрипции выполняет обратный комплемент (TCAG → CUGA), чтобы получить мРНК, рассматривая ДНК как матричную цепь. Однако в биоинформатике и так далее в Biopython мы обычно работаем напрямую с кодирующей цепью и можем получить последовательность мРНК, изменив букву Т на U.
Простой пример для вышесказанного выглядит следующим образом —
>>> from Bio.Seq import Seq >>> from Bio.Seq import transcribe >>> from Bio.Alphabet import IUPAC >>> dna_seq = Seq("ATGCCGATCGTAT",IUPAC.unambiguous_dna) >>> transcribe(dna_seq) Seq('AUGCCGAUCGUAU', IUPACUnambiguousRNA()) >>>
Чтобы изменить транскрипцию, T изменяется на U, как показано в коде ниже —
>>> rna_seq = transcribe(dna_seq) >>> rna_seq.back_transcribe() Seq('ATGCCGATCGTAT', IUPACUnambiguousDNA())
Чтобы получить ДНК-матрицу, переверните обратно транскрибированную РНК, как показано ниже —
>>> rna_seq.back_transcribe().reverse_complement() Seq('ATACGATCGGCAT', IUPACUnambiguousDNA())
Перевод
Трансляция — это процесс трансляции последовательности РНК в последовательность белка. Рассмотрим последовательность РНК, как показано ниже —
>>> rna_seq = Seq("AUGGCCAUUGUAAU",IUPAC.unambiguous_rna) >>> rna_seq Seq('AUGGCCAUUGUAAUGGGCCGCUGAAAGGGUGCCCGAUAG', IUPACUnambiguousRNA())
Теперь примените функцию translate () к коду выше —
>>> rna_seq.translate() Seq('MAIV', IUPACProtein())
Вышеуказанная последовательность РНК проста. Рассмотрим последовательность РНК, AUGGCCAUUGUAAUGGGCCGCUGAAAGGGUGCCCGA и примените translate () —
>>> rna = Seq('AUGGCCAUUGUAAUGGGCCGCUGAAAGGGUGCCCGA', IUPAC.unambiguous_rna) >>> rna.translate() Seq('MAIVMGR*KGAR', HasStopCodon(IUPACProtein(), '*'))
Здесь стоп-кодоны обозначены звездочкой «*».
В методе translate () возможно остановиться на первом стоп-кодоне. Для этого вы можете назначить to_stop = True в translate () следующим образом:
>>> rna.translate(to_stop = True) Seq('MAIVMGR', IUPACProtein())
Здесь стоп-кодон не включен в результирующую последовательность, потому что он не содержит такового.
Таблица перевода
На странице Генетических кодов NCBI представлен полный список таблиц перевода, используемых Biopython. Давайте посмотрим пример стандартной таблицы для визуализации кода —
>>> from Bio.Data import CodonTable >>> table = CodonTable.unambiguous_dna_by_name["Standard"] >>> print(table) Table 1 Standard, SGC0 | T | C | A | G | --+---------+---------+---------+---------+-- T | TTT F | TCT S | TAT Y | TGT C | T T | TTC F | TCC S | TAC Y | TGC C | C T | TTA L | TCA S | TAA Stop| TGA Stop| A T | TTG L(s)| TCG S | TAG Stop| TGG W | G --+---------+---------+---------+---------+-- C | CTT L | CCT P | CAT H | CGT R | T C | CTC L | CCC P | CAC H | CGC R | C C | CTA L | CCA P | CAA Q | CGA R | A C | CTG L(s)| CCG P | CAG Q | CGG R | G --+---------+---------+---------+---------+-- A | ATT I | ACT T | AAT N | AGT S | T A | ATC I | ACC T | AAC N | AGC S | C A | ATA I | ACA T | AAA K | AGA R | A A | ATG M(s)| ACG T | AAG K | AGG R | G --+---------+---------+---------+---------+-- G | GTT V | GCT A | GAT D | GGT G | T G | GTC V | GCC A | GAC D | GGC G | C G | GTA V | GCA A | GAA E | GGA G | A G | GTG V | GCG A | GAG E | GGG G | G --+---------+---------+---------+---------+-- >>>
Biopython использует эту таблицу для перевода ДНК в белок, а также для поиска стоп-кодона.
Biopython — последовательность операций ввода-вывода
Biopython предоставляет модуль Bio.SeqIO для чтения и записи последовательностей из и в файл (любой поток) соответственно. Он поддерживает практически все форматы файлов, доступные в биоинформатике. Большая часть программного обеспечения обеспечивает разный подход для разных форматов файлов. Но Biopython сознательно следует единому подходу, чтобы представить данные проанализированной последовательности пользователю через его объект SeqRecord.
Давайте узнаем больше о SeqRecord в следующем разделе.
SeqRecord
Модуль Bio.SeqRecord предоставляет SeqRecord для хранения мета-информации о последовательности, а также о самих данных последовательности, как показано ниже —
-
seq — это фактическая последовательность
-
id — это первичный идентификатор данной последовательности. Тип по умолчанию — строка.
-
name — это имя последовательности. Тип по умолчанию — строка.
-
description — отображает удобочитаемую информацию о последовательности.
-
аннотации — это словарь дополнительной информации о последовательности.
seq — это фактическая последовательность
id — это первичный идентификатор данной последовательности. Тип по умолчанию — строка.
name — это имя последовательности. Тип по умолчанию — строка.
description — отображает удобочитаемую информацию о последовательности.
аннотации — это словарь дополнительной информации о последовательности.
SeqRecord может быть импортирован как указано ниже
from Bio.SeqRecord import SeqRecord
Давайте разберемся в нюансах разбора файла последовательности с использованием файла реальной последовательности в следующих разделах.
Форматы файла последовательности разбора
В этом разделе объясняется, как анализировать два наиболее популярных формата файлов последовательности, FASTA и GenBank .
FASTA
FASTA — это самый основной формат файла для хранения данных последовательности. Изначально FASTA — это программный пакет для выравнивания последовательностей ДНК и белка, разработанный в начале ранней эволюции биоинформатики и использовавшийся в основном для поиска сходства последовательностей.
Biopython предоставляет пример файла FASTA, доступ к которому можно получить по адресу https://github.com/biopython/biopython/blob/master/Doc/examples/ls_orchid.fasta.
Загрузите и сохраните этот файл в каталоге образцов Biopython под именем ‘orchid.fasta’ .
Модуль Bio.SeqIO предоставляет метод parse () для обработки файлов последовательности и может быть импортирован следующим образом:
from Bio.SeqIO import parse
Метод parse () содержит два аргумента, первый — дескриптор файла, а второй — формат файла.
>>> file = open('path/to/biopython/sample/orchid.fasta') >>> for record in parse(file, "fasta"): ... print(record.id) ... gi|2765658|emb|Z78533.1|CIZ78533 gi|2765657|emb|Z78532.1|CCZ78532 .......... .......... gi|2765565|emb|Z78440.1|PPZ78440 gi|2765564|emb|Z78439.1|PBZ78439 >>>
Здесь метод parse () возвращает итеративный объект, который возвращает SeqRecord на каждой итерации. Будучи повторяемым, он предоставляет множество сложных и простых методов и позволяет нам увидеть некоторые из особенностей.
следующий()
Метод next () возвращает следующий элемент, доступный в итерируемом объекте, который мы можем использовать для получения первой последовательности, как показано ниже:
>>> first_seq_record = next(SeqIO.parse(open('path/to/biopython/sample/orchid.fasta'),'fasta')) >>> first_seq_record.id 'gi|2765658|emb|Z78533.1|CIZ78533' >>> first_seq_record.name 'gi|2765658|emb|Z78533.1|CIZ78533' >>> first_seq_record.seq Seq('CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGATGAGACCGTGG...CGC', SingleLetterAlphabet()) >>> first_seq_record.description 'gi|2765658|emb|Z78533.1|CIZ78533 C.irapeanum 5.8S rRNA gene and ITS1 and ITS2 DNA' >>> first_seq_record.annotations {} >>>
Здесь seq_record.annotations пуст, поскольку формат FASTA не поддерживает аннотации последовательности.
понимание списка
Мы можем преобразовать итерируемый объект в список, используя понимание списка, как указано ниже
>>> seq_iter = SeqIO.parse(open('path/to/biopython/sample/orchid.fasta'),'fasta') >>> all_seq = [seq_record for seq_record in seq_iter] >>> len(all_seq) 94 >>>
Здесь мы использовали метод len, чтобы получить общее количество. Мы можем получить последовательность с максимальной длиной следующим образом:
>>> seq_iter = SeqIO.parse(open('path/to/biopython/sample/orchid.fasta'),'fasta') >>> max_seq = max(len(seq_record.seq) for seq_record in seq_iter) >>> max_seq 789 >>>
Мы можем отфильтровать последовательность, используя приведенный ниже код —
>>> seq_iter = SeqIO.parse(open('path/to/biopython/sample/orchid.fasta'),'fasta') >>> seq_under_600 = [seq_record for seq_record in seq_iter if len(seq_record.seq) < 600] >>> for seq in seq_under_600: ... print(seq.id) ... gi|2765606|emb|Z78481.1|PIZ78481 gi|2765605|emb|Z78480.1|PGZ78480 gi|2765601|emb|Z78476.1|PGZ78476 gi|2765595|emb|Z78470.1|PPZ78470 gi|2765594|emb|Z78469.1|PHZ78469 gi|2765564|emb|Z78439.1|PBZ78439 >>>
Записать коллекцию объектов SqlRecord (проанализированные данные) в файл так же просто, как вызвать метод SeqIO.write, как показано ниже:
file = open("converted.fasta", "w) SeqIO.write(seq_record, file, "fasta")
Этот метод может быть эффективно использован для преобразования формата, как указано ниже —
file = open("converted.gbk", "w) SeqIO.write(seq_record, file, "genbank")
GenBank
Это более богатый формат последовательности для генов и включает поля для различных видов аннотаций. Biopython предоставляет пример файла GenBank, доступ к которому можно получить по адресу https://github.com/biopython/biopython/blob/master/Doc/examples/ls_orchid.fasta.
Загрузите и сохраните файл в вашем каталоге образцов Biopython под именем ‘orchid.gbk’
Поскольку Biopython предоставляет единственную функцию, разбирать для разбора всех форматов биоинформатики. Разбор формата GenBank так же прост, как изменение параметра формата в методе разбора.
Код для того же самого был дан ниже —
>>> from Bio import SeqIO >>> from Bio.SeqIO import parse >>> seq_record = next(parse(open('path/to/biopython/sample/orchid.gbk'),'genbank')) >>> seq_record.id 'Z78533.1' >>> seq_record.name 'Z78533' >>> seq_record.seq Seq('CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGATGAGACCGTGG...CGC', IUPACAmbiguousDNA()) >>> seq_record.description 'C.irapeanum 5.8S rRNA gene and ITS1 and ITS2 DNA' >>> seq_record.annotations { 'molecule_type': 'DNA', 'topology': 'linear', 'data_file_division': 'PLN', 'date': '30-NOV-2006', 'accessions': ['Z78533'], 'sequence_version': 1, 'gi': '2765658', 'keywords': ['5.8S ribosomal RNA', '5.8S rRNA gene', 'internal transcribed spacer', 'ITS1', 'ITS2'], 'source': 'Cypripedium irapeanum', 'organism': 'Cypripedium irapeanum', 'taxonomy': [ 'Eukaryota', 'Viridiplantae', 'Streptophyta', 'Embryophyta', 'Tracheophyta', 'Spermatophyta', 'Magnoliophyta', 'Liliopsida', 'Asparagales', 'Orchidaceae', 'Cypripedioideae', 'Cypripedium'], 'references': [ Reference(title = 'Phylogenetics of the slipper orchids (Cypripedioideae: Orchidaceae): nuclear rDNA ITS sequences', ...), Reference(title = 'Direct Submission', ...) ] }
Biopython — выравнивание последовательностей
Выравнивание последовательностей представляет собой процесс расположения двух или более последовательностей (последовательностей ДНК, РНК или белка) в определенном порядке, чтобы идентифицировать область сходства между ними.
Идентификация подобного региона позволяет нам вывести много информации, например, какие признаки сохраняются между видами, насколько генетически близки различные виды, как эволюционируют виды и т. Д. Biopython предоставляет обширную поддержку для выравнивания последовательностей.
Давайте изучим некоторые важные функции, предоставляемые Biopython в этой главе:
Выравнивание последовательности парсинга
Biopython предоставляет модуль Bio.AlignIO для чтения и записи выравниваний последовательностей. В биоинформатике существует множество доступных форматов для указания данных выравнивания последовательностей, аналогичных ранее изученным данным последовательностей. Bio.AlignIO предоставляет API, аналогичный Bio.SeqIO, за исключением того, что Bio.SeqIO работает с данными последовательности, а Bio.AlignIO работает с данными выравнивания последовательности.
Прежде чем приступить к изучению, давайте загрузим образец файла выравнивания последовательности из Интернета.
Чтобы загрузить файл примера, выполните следующие действия:
Шаг 1 — Откройте ваш любимый браузер и перейдите на сайт http://pfam.xfam.org/family/browse . Он покажет все семейства Pfam в алфавитном порядке.
Шаг 2 — Выберите любую семью с меньшим количеством семян. Он содержит минимальные данные и позволяет нам легко работать с выравниванием. Здесь мы выбрали / щелкнули PF18225, и он открывается, перейдите по адресу http://pfam.xfam.org/family/PF18225 и показывает полную информацию о нем, включая выравнивание последовательностей.
Шаг 3 — Перейдите в раздел выравнивания и загрузите файл выравнивания последовательности в формате Стокгольма (PF18225_seed.txt).
Давайте попробуем прочитать загруженный файл выравнивания последовательности, используя Bio.AlignIO, как показано ниже:
Импортировать модуль Bio.AlignIO
>>> from Bio import AlignIO
Считать выравнивание, используя метод чтения. Метод read используется для чтения отдельных данных выравнивания, доступных в данном файле. Если данный файл содержит много выравниваний, мы можем использовать метод разбора. Метод parse возвращает итеративный объект выравнивания, аналогичный методу parse в модуле Bio.SeqIO.
>>> alignment = AlignIO.read(open("PF18225_seed.txt"), "stockholm")
Распечатать объект выравнивания.
>>> print(alignment) SingleLetterAlphabet() alignment with 6 rows and 65 columns MQNTPAERLPAIIEKAKSKHDINVWLLDRQGRDLLEQRVPAKVA...EGP B7RZ31_9GAMM/59-123 AKQRGIAGLEEWLHRLDHSEAIPIFLIDEAGKDLLEREVPADIT...KKP A0A0C3NPG9_9PROT/58-119 ARRHGQEYFQQWLERQPKKVKEQVFAVDQFGRELLGRPLPEDMA...KKP A0A143HL37_9GAMM/57-121 TRRHGPESFRFWLERQPVEARDRIYAIDRSGAEILDRPIPRGMA...NKP A0A0X3UC67_9GAMM/57-121 AINRNTQQLTQDLRAMPNWSLRFVYIVDRNNQDLLKRPLPPGIM...NRK B3PFT7_CELJU/62-126 AVNATEREFTERIRTLPHWARRNVFVLDSQGFEIFDRELPSPVA...NRT K4KEM7_SIMAS/61-125 >>>
Мы также можем проверить последовательности (SeqRecord), доступные в выравнивании, а также ниже —
>>> for align in alignment: ... print(align.seq) ... MQNTPAERLPAIIEKAKSKHDINVWLLDRQGRDLLEQRVPAKVATVANQLRGRKRRAFARHREGP AKQRGIAGLEEWLHRLDHSEAIPIFLIDEAGKDLLEREVPADITA---RLDRRREHGEHGVRKKP ARRHGQEYFQQWLERQPKKVKEQVFAVDQFGRELLGRPLPEDMAPMLIALNYRNRESHAQVDKKP TRRHGPESFRFWLERQPVEARDRIYAIDRSGAEILDRPIPRGMAPLFKVLSFRNREDQGLVNNKP AINRNTQQLTQDLRAMPNWSLRFVYIVDRNNQDLLKRPLPPGIMVLAPRLTAKHPYDKVQDRNRK AVNATEREFTERIRTLPHWARRNVFVLDSQGFEIFDRELPSPVADLMRKLDLDRPFKKLERKNRT >>>
Несколько выравниваний
Как правило, большинство файлов выравнивания последовательности содержат отдельные данные выравнивания, и для их анализа достаточно использовать метод read . В концепции множественного выравнивания последовательностей две или более последовательностей сравниваются для лучшего совпадения подпоследовательностей между ними и приводит к выравниванию множественных последовательностей в одном файле.
Если формат выравнивания входной последовательности содержит более одного выравнивания последовательности, то нам нужно использовать метод разбора вместо метода чтения, как указано ниже —
>>> from Bio import AlignIO >>> alignments = AlignIO.parse(open("PF18225_seed.txt"), "stockholm") >>> print(alignments) <generator object parse at 0x000001CD1C7E0360> >>> for alignment in alignments: ... print(alignment) ... SingleLetterAlphabet() alignment with 6 rows and 65 columns MQNTPAERLPAIIEKAKSKHDINVWLLDRQGRDLLEQRVPAKVA...EGP B7RZ31_9GAMM/59-123 AKQRGIAGLEEWLHRLDHSEAIPIFLIDEAGKDLLEREVPADIT...KKP A0A0C3NPG9_9PROT/58-119 ARRHGQEYFQQWLERQPKKVKEQVFAVDQFGRELLGRPLPEDMA...KKP A0A143HL37_9GAMM/57-121 TRRHGPESFRFWLERQPVEARDRIYAIDRSGAEILDRPIPRGMA...NKP A0A0X3UC67_9GAMM/57-121 AINRNTQQLTQDLRAMPNWSLRFVYIVDRNNQDLLKRPLPPGIM...NRK B3PFT7_CELJU/62-126 AVNATEREFTERIRTLPHWARRNVFVLDSQGFEIFDRELPSPVA...NRT K4KEM7_SIMAS/61-125 >>>
Здесь метод parse возвращает итеративный объект выравнивания, и его можно повторять, чтобы получить фактические выравнивания.
Парное выравнивание последовательности
Попарное выравнивание последовательностей сравнивает только две последовательности одновременно и обеспечивает наилучшие возможные выравнивания последовательностей. Попарно легко понять и исключить из полученного выравнивания последовательности.
Biopython предоставляет специальный модуль Bio.pairwise2 для идентификации последовательности выравнивания с использованием попарного метода. Biopython применяет лучший алгоритм, чтобы найти последовательность выравнивания, и это то же самое, что и другое программное обеспечение.
Напишем пример, чтобы найти выравнивание последовательностей двух простых и гипотетических последовательностей с использованием попарного модуля. Это поможет нам понять концепцию выравнивания последовательностей и как ее запрограммировать с помощью Biopython.
Шаг 1
Импортируйте модуль pairwise2 с помощью команды, приведенной ниже —
>>> from Bio import pairwise2
Шаг 2
Создайте две последовательности, seq1 и seq2 —
>>> from Bio.Seq import Seq >>> seq1 = Seq("ACCGGT") >>> seq2 = Seq("ACGT")
Шаг 3
Вызовите метод pairwise2.align.globalxx вместе с seq1 и seq2, чтобы найти выравнивания, используя строку кода ниже —
>>> alignments = pairwise2.align.globalxx(seq1, seq2)
Здесь метод globalxx выполняет фактическую работу и находит все наилучшие возможные выравнивания в заданных последовательностях. На самом деле, Bio.pairwise2 предоставляет целый набор методов, которые следуют приведенному ниже соглашению, чтобы найти выравнивания в различных сценариях.
<sequence alignment type>XY
Здесь тип выравнивания последовательности относится к типу выравнивания, который может быть глобальным или локальным. глобальный тип находит выравнивание последовательности, принимая во внимание всю последовательность. Локальный тип находит выравнивание последовательностей, просматривая также подмножество заданных последовательностей. Это будет утомительно, но дает лучшее представление о сходстве данных последовательностей.
-
Х относится к совпадению баллов. Возможные значения: x (точное совпадение), m (оценка, основанная на одинаковых символах), d (пользовательский словарь с символом и счетом совпадения) и, наконец, c (определенная пользователем функция для предоставления собственного алгоритма оценки).
-
Y относится к штрафу за разрыв. Возможные значения: x (без штрафных санкций), s (одинаковые штрафы для обеих последовательностей), d (разные штрафы для каждой последовательности) и, наконец, c (пользовательская функция для предоставления пользовательских штрафов за пропуск)
Х относится к совпадению баллов. Возможные значения: x (точное совпадение), m (оценка, основанная на одинаковых символах), d (пользовательский словарь с символом и счетом совпадения) и, наконец, c (определенная пользователем функция для предоставления собственного алгоритма оценки).
Y относится к штрафу за разрыв. Возможные значения: x (без штрафных санкций), s (одинаковые штрафы для обеих последовательностей), d (разные штрафы для каждой последовательности) и, наконец, c (пользовательская функция для предоставления пользовательских штрафов за пропуск)
Таким образом, localds также является допустимым методом, который находит выравнивание последовательности, используя технику локального выравнивания, пользовательский словарь для совпадений и пользовательский штраф за пробел для обеих последовательностей.
>>> test_alignments = pairwise2.align.localds(seq1, seq2, blosum62, -10, -1)
Здесь blosum62 относится к словарю, доступному в модуле pairwise2, для обеспечения соответствия. -10 относится к штрафу за открытие пропуска, а -1 относится к штрафу за расширение пропуска.
Шаг 4
Зациклите итеративный объект выравнивания и получите каждый отдельный объект выравнивания и распечатайте его.
>>> for alignment in alignments:
... print(alignment)
...
('ACCGGT', 'A-C-GT', 4.0, 0, 6)
('ACCGGT', 'AC--GT', 4.0, 0, 6)
('ACCGGT', 'A-CG-T', 4.0, 0, 6)
('ACCGGT', 'AC-G-T', 4.0, 0, 6)
Шаг 5
Модуль Bio.pairwise2 предоставляет метод форматирования format_alignment для лучшей визуализации результата —
>>> from Bio.pairwise2 import format_alignment >>> alignments = pairwise2.align.globalxx(seq1, seq2) >>> for alignment in alignments: ... print(format_alignment(*alignment)) ... ACCGGT | | || A-C-GT Score=4 ACCGGT || || AC--GT Score=4 ACCGGT | || | A-CG-T Score=4 ACCGGT || | | AC-G-T Score=4 >>>
Biopython также предоставляет другой модуль для выравнивания последовательностей, Align. Этот модуль предоставляет другой набор API для простой установки параметров, таких как алгоритм, режим, оценка совпадения, штрафы за пропуски и т. Д. Простой взгляд на объект Align выглядит следующим образом:
>>> from Bio import Align >>> aligner = Align.PairwiseAligner() >>> print(aligner) Pairwise sequence aligner with parameters match score: 1.000000 mismatch score: 0.000000 target open gap score: 0.000000 target extend gap score: 0.000000 target left open gap score: 0.000000 target left extend gap score: 0.000000 target right open gap score: 0.000000 target right extend gap score: 0.000000 query open gap score: 0.000000 query extend gap score: 0.000000 query left open gap score: 0.000000 query left extend gap score: 0.000000 query right open gap score: 0.000000 query right extend gap score: 0.000000 mode: global >>>
Поддержка инструментов выравнивания последовательностей
Biopython предоставляет интерфейс для многих инструментов выравнивания последовательностей через модуль Bio.Align.Applications. Некоторые из инструментов перечислены ниже —
- ClustalW
- MUSCLE
- EMBOSS игла и вода
Давайте напишем простой пример в Biopython для создания выравнивания последовательностей с помощью самого популярного инструмента выравнивания ClustalW.
Шаг 1 — Загрузите программу Clustalw с http://www.clustal.org/download/current/ и установите ее. Также обновите системный PATH, указав путь установки «clustal».
Шаг 2 — импортировать ClustalwCommanLine из модуля Bio.Align.Applications.
>>> from Bio.Align.Applications import ClustalwCommandline
Шаг 3 — Установите cmd, вызвав ClustalwCommanLine с входным файлом, opuntia.fasta, доступным в пакете Biopython. https://raw.githubusercontent.com/biopython/biopython/master/Doc/examples/opuntia.fasta
>>> cmd = ClustalwCommandline("clustalw2",
infile="/path/to/biopython/sample/opuntia.fasta")
>>> print(cmd)
clustalw2 -infile=fasta/opuntia.fasta
Шаг 4 — вызов cmd () запустит команду clustalw и выдаст выходной файл результирующего выравнивания opuntia.aln.
>>> stdout, stderr = cmd()
Шаг 5 — Прочитайте и распечатайте файл выравнивания, как показано ниже —
>>> from Bio import AlignIO
>>> align = AlignIO.read("/path/to/biopython/sample/opuntia.aln", "clustal")
>>> print(align)
SingleLetterAlphabet() alignment with 7 rows and 906 columns
TATACATTAAAGAAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273285|gb|AF191659.1|AF191
TATACATTAAAGAAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273284|gb|AF191658.1|AF191
TATACATTAAAGAAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273287|gb|AF191661.1|AF191
TATACATAAAAGAAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273286|gb|AF191660.1|AF191
TATACATTAAAGGAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273290|gb|AF191664.1|AF191
TATACATTAAAGGAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273289|gb|AF191663.1|AF191
TATACATTAAAGGAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA
gi|6273291|gb|AF191665.1|AF191
>>>
Биопайтон — обзор BLAST
BLAST расшифровывается как Basic Local Alignment Tool . Он находит области сходства между биологическими последовательностями. Biopython предоставляет модуль Bio.Blast для работы с NCBI BLAST. Вы можете запустить BLAST в локальном или интернет-соединении.
Давайте кратко разберемся в этих двух связях в следующем разделе —
Работает через интернет
Biopython предоставляет модуль Bio.Blast.NCBIWWW для вызова онлайн-версии BLAST. Для этого нам нужно импортировать следующий модуль —
>>> from Bio.Blast import NCBIWWW
Модуль NCBIWW предоставляет функцию qblast для запроса онлайн-версии BLAST, https://blast.ncbi.nlm.nih.gov/Blast.cgi . qblast поддерживает все параметры, поддерживаемые онлайн-версией.
Чтобы получить какую-либо справку об этом модуле, используйте приведенную ниже команду и поймите функции —
>>> help(NCBIWWW.qblast) Help on function qblast in module Bio.Blast.NCBIWWW: qblast( program, database, sequence, url_base = 'https://blast.ncbi.nlm.nih.gov/Blast.cgi', auto_format = None, composition_based_statistics = None, db_genetic_code = None, endpoints = None, entrez_query = '(none)', expect = 10.0, filter = None, gapcosts = None, genetic_code = None, hitlist_size = 50, i_thresh = None, layout = None, lcase_mask = None, matrix_name = None, nucl_penalty = None, nucl_reward = None, other_advanced = None, perc_ident = None, phi_pattern = None, query_file = None, query_believe_defline = None, query_from = None, query_to = None, searchsp_eff = None, service = None, threshold = None, ungapped_alignment = None, word_size = None, alignments = 500, alignment_view = None, descriptions = 500, entrez_links_new_window = None, expect_low = None, expect_high = None, format_entrez_query = None, format_object = None, format_type = 'XML', ncbi_gi = None, results_file = None, show_overview = None, megablast = None, template_type = None, template_length = None ) BLAST search using NCBI's QBLAST server or a cloud service provider. Supports all parameters of the qblast API for Put and Get. Please note that BLAST on the cloud supports the NCBI-BLAST Common URL API (http://ncbi.github.io/blast-cloud/dev/api.html). To use this feature, please set url_base to 'http://host.my.cloud.service.provider.com/cgi-bin/blast.cgi' and format_object = 'Alignment'. For more details, please see 8. Biopython – Overview of BLAST https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE = BlastDocs&DOC_TYPE = CloudBlast Some useful parameters: - program blastn, blastp, blastx, tblastn, or tblastx (lower case) - database Which database to search against (e.g. "nr"). - sequence The sequence to search. - ncbi_gi TRUE/FALSE whether to give 'gi' identifier. - descriptions Number of descriptions to show. Def 500. - alignments Number of alignments to show. Def 500. - expect An expect value cutoff. Def 10.0. - matrix_name Specify an alt. matrix (PAM30, PAM70, BLOSUM80, BLOSUM45). - filter "none" turns off filtering. Default no filtering - format_type "HTML", "Text", "ASN.1", or "XML". Def. "XML". - entrez_query Entrez query to limit Blast search - hitlist_size Number of hits to return. Default 50 - megablast TRUE/FALSE whether to use MEga BLAST algorithm (blastn only) - service plain, psi, phi, rpsblast, megablast (lower case) This function does no checking of the validity of the parameters and passes the values to the server as is. More help is available at: https://ncbi.github.io/blast-cloud/dev/api.html
Обычно аргументы функции qblast в основном аналогичны различным параметрам, которые вы можете установить на веб-странице BLAST. Это облегчает понимание функции qblast, а также сокращает кривую обучения ее использованию.
Подключение и поиск
Чтобы понять процесс подключения и поиска в онлайн-версии BLAST, давайте выполним простой поиск последовательностей (доступен в нашем локальном файле последовательностей) на онлайн-сервере BLAST через Biopython.
Шаг 1 — Создайте файл с именем blast_example.fasta в каталоге Biopython и передайте приведенную ниже информацию о последовательности в качестве входных данных.
Example of a single sequence in FASTA/Pearson format: >sequence A ggtaagtcctctagtacaaacacccccaatattgtgatataattaaaattatattcatat tctgttgccagaaaaaacacttttaggctatattagagccatcttctttgaagcgttgtc >sequence B ggtaagtcctctagtacaaacacccccaatattgtgatataattaaaattatattca tattctgttgccagaaaaaacacttttaggctatattagagccatcttctttgaagcgttgtc
Шаг 2 — Импортируйте модуль NCBIWWW.
>>> from Bio.Blast import NCBIWWW
Шаг 3 — Откройте файл последовательности blast_example.fasta с помощью модуля ввода-вывода Python.
>>> sequence_data = open("blast_example.fasta").read()
>>> sequence_data
'Example of a single sequence in FASTA/Pearson format:\n\n\n> sequence
A\nggtaagtcctctagtacaaacacccccaatattgtgatataattaaaatt
atattcatat\ntctgttgccagaaaaaacacttttaggctatattagagccatcttctttg aagcgttgtc\n\n'
Шаг 4 — Теперь вызовите функцию qblast, передавая данные последовательности в качестве основного параметра. Другой параметр представляет базу данных (nt) и внутреннюю программу (blastn).
>>> result_handle = NCBIWWW.qblast("blastn", "nt", sequence_data)
>>> result_handle
<_io.StringIO object at 0x000001EC9FAA4558>
blast_results содержит результат нашего поиска. Он может быть сохранен в файл для последующего использования, а также проанализирован для получения подробной информации. Мы узнаем, как это сделать, в следующем разделе.
Шаг 5 — Те же функциональные возможности могут быть выполнены с использованием объекта Seq, а не с использованием всего файла fasta, как показано ниже —
>>> from Bio import SeqIO
>>> seq_record = next(SeqIO.parse(open('blast_example.fasta'),'fasta'))
>>> seq_record.id
'sequence'
>>> seq_record.seq
Seq('ggtaagtcctctagtacaaacacccccaatattgtgatataattaaaattatat...gtc',
SingleLetterAlphabet())
Теперь вызовите функцию qblast, передав объект Seq, record.seq в качестве основного параметра.
>>> result_handle = NCBIWWW.qblast("blastn", "nt", seq_record.seq)
>>> print(result_handle)
<_io.StringIO object at 0x000001EC9FAA4558>
BLAST автоматически назначит идентификатор вашей последовательности.
Шаг 6 — объект result_handle будет иметь весь результат и может быть сохранен в файл для дальнейшего использования.
>>> with open('results.xml', 'w') as save_file:
>>> blast_results = result_handle.read()
>>> save_file.write(blast_results)
Мы увидим, как разобрать файл результатов в следующем разделе.
Запуск автономного BLAST
В этом разделе объясняется, как запустить BLAST в локальной системе. Если вы запускаете BLAST в локальной системе, это может быть быстрее, а также позволяет создавать собственную базу данных для поиска по последовательностям.
Подключение BLAST
В общем, запуск BLAST локально не рекомендуется из-за его большого размера, дополнительных усилий, необходимых для запуска программного обеспечения, и связанных с этим затрат. Онлайн BLAST достаточно для базовых и продвинутых целей. Конечно, иногда вам может потребоваться установить его локально.
Предположим, что вы проводите частые поиски в Интернете, что может потребовать много времени и большого объема сети, и если у вас имеются запатентованные данные последовательности или проблемы, связанные с IP, рекомендуется установить его локально.
Для этого нам нужно выполнить следующие шаги —
Шаг 1 — Загрузите и установите последний двоичный файл Blast, используя данную ссылку — ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/
Шаг 2 — Загрузите и распакуйте последнюю и необходимую базу данных, используя ссылку ниже — ftp://ftp.ncbi.nlm.nih.gov/blast/db/
Программное обеспечение BLAST предоставляет множество баз данных на своем сайте. Давайте загрузим файл alu.n.gz с сайта базы данных Blast и распакуем его в папку alu. Этот файл в формате FASTA. Чтобы использовать этот файл в нашем приложении Blast, нам нужно сначала преобразовать файл из формата FASTA в формат базы данных Blast. BLAST предоставляет приложение makeblastdb для этого преобразования.
Используйте приведенный ниже фрагмент кода —
cd /path/to/alu makeblastdb -in alu.n -parse_seqids -dbtype nucl -out alun
Выполнение приведенного выше кода проанализирует входной файл alu.n и создаст базу данных BLAST как несколько файлов alun.nsq, alun.nsi и т. Д. Теперь мы можем запросить эту базу данных, чтобы найти последовательность.
Мы установили BLAST на наш локальный сервер, а также имеем образец базы данных BLAST, позволяющий выполнять запросы к нему.
Шаг 3 — Давайте создадим пример файла последовательности для запроса базы данных. Создайте файл search.fsa и поместите в него приведенные ниже данные.
>gnl|alu|Z15030_HSAL001056 (Alu-J) AGGCTGGCACTGTGGCTCATGCTGAAATCCCAGCACGGCGGAGGACGGCGGAAGATTGCT TGAGCCTAGGAGTTTGCGACCAGCCTGGGTGACATAGGGAGATGCCTGTCTCTACGCAAA AGAAAAAAAAAATAGCTCTGCTGGTGGTGCATGCCTATAGTCTCAGCTATCAGGAGGCTG GGACAGGAGGATCACTTGGGCCCGGGAGTTGAGGCTGTGGTGAGCCACGATCACACCACT GCACTCCAGCCTGGGTGACAGAGCAAGACCCTGTCTCAAAACAAACAAATAA >gnl|alu|D00596_HSAL003180 (Alu-Sx) AGCCAGGTGTGGTGGCTCACGCCTGTAATCCCACCGCTTTGGGAGGCTGAGTCAGATCAC CTGAGGTTAGGAATTTGGGACCAGCCTGGCCAACATGGCGACACCCCAGTCTCTACTAAT AACACAAAAAATTAGCCAGGTGTGCTGGTGCATGTCTGTAATCCCAGCTACTCAGGAGGC TGAGGCATGAGAATTGCTCACGAGGCGGAGGTTGTAGTGAGCTGAGATCGTGGCACTGTA CTCCAGCCTGGCGACAGAGGGAGAACCCATGTCAAAAACAAAAAAAGACACCACCAAAGG TCAAAGCATA >gnl|alu|X55502_HSAL000745 (Alu-J) TGCCTTCCCCATCTGTAATTCTGGCACTTGGGGAGTCCAAGGCAGGATGATCACTTATGC CCAAGGAATTTGAGTACCAAGCCTGGGCAATATAACAAGGCCCTGTTTCTACAAAAACTT TAAACAATTAGCCAGGTGTGGTGGTGCGTGCCTGTGTCCAGCTACTCAGGAAGCTGAGGC AAGAGCTTGAGGCTACAGTGAGCTGTGTTCCACCATGGTGCTCCAGCCTGGGTGACAGGG CAAGACCCTGTCAAAAGAAAGGAAGAAAGAACGGAAGGAAAGAAGGAAAGAAACAAGGAG AG
Данные о последовательности собраны из файла alu.n; следовательно, это соответствует нашей базе данных.
Шаг 4 — Программное обеспечение BLAST предоставляет множество приложений для поиска в базе данных, и мы используем blastn. Приложение blastn требует минимум трех аргументов: db, query и out. дБ относится к базе данных против поиска; запрос — это последовательность для сравнения, а файл для сохранения результатов. Теперь выполните следующую команду для выполнения этого простого запроса:
blastn -db alun -query search.fsa -out results.xml -outfmt 5
Выполнение вышеуказанной команды произведет поиск и выдаст вывод в файле results.xml, как показано ниже (частично данные) —
<?xml version = "1.0"?>
<!DOCTYPE BlastOutput PUBLIC "-//NCBI//NCBI BlastOutput/EN"
"http://www.ncbi.nlm.nih.gov/dtd/NCBI_BlastOutput.dtd">
<BlastOutput>
<BlastOutput_program>blastn</BlastOutput_program>
<BlastOutput_version>BLASTN 2.7.1+</BlastOutput_version>
<BlastOutput_reference>Zheng Zhang, Scott Schwartz, Lukas Wagner, and Webb
Miller (2000), "A greedy algorithm for aligning DNA sequences", J
Comput Biol 2000; 7(1-2):203-14.
</BlastOutput_reference>
<BlastOutput_db>alun</BlastOutput_db>
<BlastOutput_query-ID>Query_1</BlastOutput_query-ID>
<BlastOutput_query-def>gnl|alu|Z15030_HSAL001056 (Alu-J)</BlastOutput_query-def>
<BlastOutput_query-len>292</BlastOutput_query-len>
<BlastOutput_param>
<Parameters>
<Parameters_expect>10</Parameters_expect>
<Parameters_sc-match>1</Parameters_sc-match>
<Parameters_sc-mismatch>-2</Parameters_sc-mismatch>
<Parameters_gap-open>0</Parameters_gap-open>
<Parameters_gap-extend>0</Parameters_gap-extend>
<Parameters_filter>L;m;</Parameters_filter>
</Parameters>
</BlastOutput_param>
<BlastOutput_iterations>
<Iteration>
<Iteration_iter-num>1</Iteration_iter-num><Iteration_query-ID>Query_1</Iteration_query-ID>
<Iteration_query-def>gnl|alu|Z15030_HSAL001056 (Alu-J)</Iteration_query-def>
<Iteration_query-len>292</Iteration_query-len>
<Iteration_hits>
<Hit>
<Hit_num>1</Hit_num>
<Hit_id>gnl|alu|Z15030_HSAL001056</Hit_id>
<Hit_def>(Alu-J)</Hit_def>
<Hit_accession>Z15030_HSAL001056</Hit_accession>
<Hit_len>292</Hit_len>
<Hit_hsps>
<Hsp>
<Hsp_num>1</Hsp_num>
<Hsp_bit-score>540.342</Hsp_bit-score>
<Hsp_score>292</Hsp_score>
<Hsp_evalue>4.55414e-156</Hsp_evalue>
<Hsp_query-from>1</Hsp_query-from>
<Hsp_query-to>292</Hsp_query-to>
<Hsp_hit-from>1</Hsp_hit-from>
<Hsp_hit-to>292</Hsp_hit-to>
<Hsp_query-frame>1</Hsp_query-frame>
<Hsp_hit-frame>1</Hsp_hit-frame>
<Hsp_identity>292</Hsp_identity>
<Hsp_positive>292</Hsp_positive>
<Hsp_gaps>0</Hsp_gaps>
<Hsp_align-len>292</Hsp_align-len>
<Hsp_qseq>
AGGCTGGCACTGTGGCTCATGCTGAAATCCCAGCACGGCGGAGGACGGCGGAAGATTGCTTGAGCCTAGGAGTTTG
CGACCAGCCTGGGTGACATAGGGAGATGCCTGTCTCTACGCAAAAGAAAAAAAAAATAGCTCTGCTGGTGGTGCATG
CCTATAGTCTCAGCTATCAGGAGGCTGGGACAGGAGGATCACTTGGGCCCGGGAGTTGAGGCTGTGGTGAGCC
ACGATCACACCACTGCACTCCAGCCTGGGTGACAGAGCAAGACCCTGTCTCAAAACAAACAAATAA
</Hsp_qseq>
<Hsp_hseq>
AGGCTGGCACTGTGGCTCATGCTGAAATCCCAGCACGGCGGAGGACGGCGGAAGATTGCTTGAGCCTAGGA
GTTTGCGACCAGCCTGGGTGACATAGGGAGATGCCTGTCTCTACGCAAAAGAAAAAAAAAATAGCTCTGCT
GGTGGTGCATGCCTATAGTCTCAGCTATCAGGAGGCTGGGACAGGAGGATCACTTGGGCCCGGGAGTTGAGG
CTGTGGTGAGCCACGATCACACCACTGCACTCCAGCCTGGGTGACAGAGCAAGACCCTGTCTCAAAACAAAC
AAATAA
</Hsp_hseq>
<Hsp_midline>
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||
</Hsp_midline>
</Hsp>
</Hit_hsps>
</Hit>
.........................
.........................
.........................
</Iteration_hits>
<Iteration_stat>
<Statistics>
<Statistics_db-num>327</Statistics_db-num>
<Statistics_db-len>80506</Statistics_db-len>
<Statistics_hsp-lenv16</Statistics_hsp-len>
<Statistics_eff-space>21528364</Statistics_eff-space>
<Statistics_kappa>0.46</Statistics_kappa>
<Statistics_lambda>1.28</Statistics_lambda>
<Statistics_entropy>0.85</Statistics_entropy>
</Statistics>
</Iteration_stat>
</Iteration>
</BlastOutput_iterations>
</BlastOutput>
Вышеприведенную команду можно запустить внутри питона, используя следующий код:
>>> from Bio.Blast.Applications import NcbiblastnCommandline >>> blastn_cline = NcbiblastnCommandline(query = "search.fasta", db = "alun", outfmt = 5, out = "results.xml") >>> stdout, stderr = blastn_cline()
Здесь первый — дескриптор взрыва, а второй — возможный вывод ошибки, сгенерированный командой взрыва.
Поскольку мы предоставили выходной файл в качестве аргумента командной строки (out = «results.xml») и установили выходной формат как XML (outfmt = 5), выходной файл будет сохранен в текущем рабочем каталоге.
Разбор BLAST Результат
Обычно вывод BLAST анализируется в формате XML с использованием модуля NCBIXML. Для этого нам нужно импортировать следующий модуль —
>>> from Bio.Blast import NCBIXML
Теперь откройте файл напрямую с помощью метода открытия Python и используйте метод разбора NCBIXML, как показано ниже —
>>> E_VALUE_THRESH = 1e-20
>>> for record in NCBIXML.parse(open("results.xml")):
>>> if record.alignments:
>>> print("\n")
>>> print("query: %s" % record.query[:100])
>>> for align in record.alignments:
>>> for hsp in align.hsps:
>>> if hsp.expect < E_VALUE_THRESH:
>>> print("match: %s " % align.title[:100])
Это произведет вывод следующим образом —
query: gnl|alu|Z15030_HSAL001056 (Alu-J) match: gnl|alu|Z15030_HSAL001056 (Alu-J) match: gnl|alu|L12964_HSAL003860 (Alu-J) match: gnl|alu|L13042_HSAL003863 (Alu-FLA?) match: gnl|alu|M86249_HSAL001462 (Alu-FLA?) match: gnl|alu|M29484_HSAL002265 (Alu-J) query: gnl|alu|D00596_HSAL003180 (Alu-Sx) match: gnl|alu|D00596_HSAL003180 (Alu-Sx) match: gnl|alu|J03071_HSAL001860 (Alu-J) match: gnl|alu|X72409_HSAL005025 (Alu-Sx) query: gnl|alu|X55502_HSAL000745 (Alu-J) match: gnl|alu|X55502_HSAL000745 (Alu-J)
Биопайтон — База данных Entrez
Entrez — это онлайновая поисковая система, предоставляемая NCBI. Он обеспечивает доступ почти ко всем известным базам данных молекулярной биологии с помощью встроенного глобального запроса, поддерживающего логические операторы и поиск по полю. Он возвращает результаты из всех баз данных с информацией, такой как количество обращений к каждой базе данных, записи со ссылками на исходную базу данных и т. Д.
Некоторые из популярных баз данных, к которым можно получить доступ через Entrez, перечислены ниже —
- Pubmed
- Изданный Центральный
- Нуклеотид (база данных последовательностей GenBank)
- Белок (база данных последовательностей)
- Геном (База данных всего генома)
- Структура (трехмерная макромолекулярная структура)
- Таксономия (Организмы в GenBank)
- SNP (однонуклеотидный полиморфизм)
- UniGene (Геноориентированные кластеры транскрипционных последовательностей)
- CDD (база данных по консервативным белковым доменам)
- 3D Домены (Домены от Entrez Structure)
В дополнение к вышеупомянутым базам данных Entrez предоставляет гораздо больше баз данных для выполнения поиска по полю.
Biopython предоставляет специальный модуль для Entrez Bio.Entrez для доступа к базе данных Entrez. Давайте узнаем, как получить доступ к Entrez с помощью Biopython в этой главе —
Шаги соединения с базой данных
Чтобы добавить функции Entrez, импортируйте следующий модуль —
>>> from Bio import Entrez
Затем установите свой адрес электронной почты, чтобы определить, кто связан с кодом, приведенным ниже —
>>> Entrez.email = '<youremail>'
Затем установите параметр инструмента Entrez и по умолчанию это Biopython.
>>> Entrez.tool = 'Demoscript'
Теперь вызовите функцию einfo, чтобы найти счетчик терминов индекса, последнее обновление и доступные ссылки для каждой базы данных, как определено ниже —
>>> info = Entrez.einfo()
Метод einfo возвращает объект, который обеспечивает доступ к информации через метод read, как показано ниже —
>>> data = info.read()
>>> print(data)
<?xml version = "1.0" encoding = "UTF-8" ?>
<!DOCTYPE eInfoResult PUBLIC "-//NLM//DTD einfo 20130322//EN"
"https://eutils.ncbi.nlm.nih.gov/eutils/dtd/20130322/einfo.dtd">
<eInfoResult>
<DbList>
<DbName>pubmed</DbName>
<DbName>protein</DbName>
<DbName>nuccore</DbName>
<DbName>ipg</DbName>
<DbName>nucleotide</DbName>
<DbName>nucgss</DbName>
<DbName>nucest</DbName>
<DbName>structure</DbName>
<DbName>sparcle</DbName>
<DbName>genome</DbName>
<DbName>annotinfo</DbName>
<DbName>assembly</DbName>
<DbName>bioproject</DbName>
<DbName>biosample</DbName>
<DbName>blastdbinfo</DbName>
<DbName>books</DbName>
<DbName>cdd</DbName>
<DbName>clinvar</DbName>
<DbName>clone</DbName>
<DbName>gap</DbName>
<DbName>gapplus</DbName>
<DbName>grasp</DbName>
<DbName>dbvar</DbName>
<DbName>gene</DbName>
<DbName>gds</DbName>
<DbName>geoprofiles</DbName>
<DbName>homologene</DbName>
<DbName>medgen</DbName>
<DbName>mesh</DbName>
<DbName>ncbisearch</DbName>
<DbName>nlmcatalog</DbName>
<DbName>omim</DbName>
<DbName>orgtrack</DbName>
<DbName>pmc</DbName>
<DbName>popset</DbName>
<DbName>probe</DbName>
<DbName>proteinclusters</DbName>
<DbName>pcassay</DbName>
<DbName>biosystems</DbName>
<DbName>pccompound</DbName>
<DbName>pcsubstance</DbName>
<DbName>pubmedhealth</DbName>
<DbName>seqannot</DbName>
<DbName>snp</DbName>
<DbName>sra</DbName>
<DbName>taxonomy</DbName>
<DbName>biocollections</DbName>
<DbName>unigene</DbName>
<DbName>gencoll</DbName>
<DbName>gtr</DbName>
</DbList>
</eInfoResult>
Данные представлены в формате XML, и для получения данных в виде объекта python используйте метод Entrez.read, как только вызывается метод Entrez.einfo () —
>>> info = Entrez.einfo() >>> record = Entrez.read(info)
Здесь record — это словарь, который имеет один ключ, DbList, как показано ниже —
>>> record.keys() [u'DbList']
Доступ к ключу DbList возвращает список имен баз данных, показанный ниже —
>>> record[u'DbList'] ['pubmed', 'protein', 'nuccore', 'ipg', 'nucleotide', 'nucgss', 'nucest', 'structure', 'sparcle', 'genome', 'annotinfo', 'assembly', 'bioproject', 'biosample', 'blastdbinfo', 'books', 'cdd', 'clinvar', 'clone', 'gap', 'gapplus', 'grasp', 'dbvar', 'gene', 'gds', 'geoprofiles', 'homologene', 'medgen', 'mesh', 'ncbisearch', 'nlmcatalog', 'omim', 'orgtrack', 'pmc', 'popset', 'probe', 'proteinclusters', 'pcassay', 'biosystems', 'pccompound', 'pcsubstance', 'pubmedhealth', 'seqannot', 'snp', 'sra', 'taxonomy', 'biocollections', 'unigene', 'gencoll', 'gtr'] >>>
По сути, модуль Entrez анализирует XML, возвращаемый поисковой системой Entrez, и предоставляет его в виде словаря и списков Python.
База данных поиска
Для поиска в любой из баз данных Entrez мы можем использовать модуль Bio.Entrez.esearch (). Это определено ниже —
>>> info = Entrez.einfo()
>>> info = Entrez.esearch(db = "pubmed",term = "genome")
>>> record = Entrez.read(info)
>>>print(record)
DictElement({u'Count': '1146113', u'RetMax': '20', u'IdList':
['30347444', '30347404', '30347317', '30347292',
'30347286', '30347249', '30347194', '30347187',
'30347172', '30347088', '30347075', '30346992',
'30346990', '30346982', '30346980', '30346969',
'30346962', '30346954', '30346941', '30346939'],
u'TranslationStack': [DictElement({u'Count':
'927819', u'Field': 'MeSH Terms', u'Term': '"genome"[MeSH Terms]',
u'Explode': 'Y'}, attributes = {})
, DictElement({u'Count': '422712', u'Field':
'All Fields', u'Term': '"genome"[All Fields]', u'Explode': 'N'}, attributes = {}),
'OR', 'GROUP'], u'TranslationSet': [DictElement({u'To': '"genome"[MeSH Terms]
OR "genome"[All Fields]', u'From': 'genome'}, attributes = {})], u'RetStart': '0',
u'QueryTranslation': '"genome"[MeSH Terms] OR "genome"[All Fields]'},
attributes = {})
>>>
Если вы назначите неправильный БД, то он возвращает
>>> info = Entrez.esearch(db = "blastdbinfo",term = "books")
>>> record = Entrez.read(info)
>>> print(record)
DictElement({u'Count': '0', u'RetMax': '0', u'IdList': [],
u'WarningList': DictElement({u'OutputMessage': ['No items found.'],
u'PhraseIgnored': [], u'QuotedPhraseNotFound': []}, attributes = {}),
u'ErrorList': DictElement({u'FieldNotFound': [], u'PhraseNotFound':
['books']}, attributes = {}), u'TranslationSet': [], u'RetStart': '0',
u'QueryTranslation': '(books[All Fields])'}, attributes = {})
Если вы хотите искать по базе данных, то вы можете использовать Entrez.egquery . Это похоже на Entrez.esearch за исключением того, что достаточно указать ключевое слово и пропустить параметр базы данных.
>>>info = Entrez.egquery(term = "entrez") >>> record = Entrez.read(info) >>> for row in record["eGQueryResult"]: ... print(row["DbName"], row["Count"]) ... pubmed 458 pmc 12779 mesh 1 ... ... ... biosample 7 biocollections 0
Fetch Records
Enterz предоставляет специальный метод efetch для поиска и загрузки полной информации о записи из Entrez. Рассмотрим следующий простой пример —
>>> handle = Entrez.efetch( db = "nucleotide", id = "EU490707", rettype = "fasta")
Теперь мы можем просто читать записи, используя объект SeqIO
>>> record = SeqIO.read( handle, "fasta" )
>>> record
SeqRecord(seq = Seq('ATTTTTTACGAACCTGTGGAAATTTTTGGTTATGACAATAAATCTAGTTTAGTA...GAA',
SingleLetterAlphabet()), id = 'EU490707.1', name = 'EU490707.1',
description = 'EU490707.1
Selenipedium aequinoctiale maturase K (matK) gene, partial cds; chloroplast',
dbxrefs = [])
Биопайтон — Модуль PDB
Biopython предоставляет модуль Bio.PDB для манипулирования полипептидными структурами. PDB (Protein Data Bank) является крупнейшим ресурсом по структуре белка, доступным онлайн. Он содержит много различных белковых структур, в том числе белково-белковые, белково-ДНК, белково-РНК-комплексы.
Чтобы загрузить PDB, введите следующую команду —
from Bio.PDB import *
Форматы файлов структуры белка
PDB распределяет белковые структуры в трех разных форматах —
- Формат файла на основе XML, который не поддерживается Biopython
- Формат файла pdb, который представляет собой специально отформатированный текстовый файл
- Формат файлов PDBx / mmCIF
Файлы PDB, распространяемые Банком Белковых Данных, могут содержать ошибки форматирования, которые делают их двусмысленными или трудными для анализа. Модуль Bio.PDB пытается автоматически устранить эти ошибки.
Модуль Bio.PDB реализует два разных анализатора, один в формате mmCIF, а второй в формате pdb.
Давайте узнаем, как анализировать каждый формат в деталях —
Парсер mmCIF
Давайте загрузим пример базы данных в формате mmCIF с сервера pdb, используя следующую команду:
>>> pdbl = PDBList()
>>> pdbl.retrieve_pdb_file('2FAT', pdir = '.', file_format = 'mmCif')
Это загрузит указанный файл (2fat.cif) с сервера и сохранит его в текущем рабочем каталоге.
Здесь PDBList предоставляет опции для просмотра и загрузки файлов с FTP-сервера PDB. Метод retrieve_pdb_file требует имя файла для загрузки без расширения. В файле retrieve_pdb_file также есть возможность указать каталог загрузки, pdir и формат файла file_format. Возможные значения формата файла следующие:
- «MmCif» (по умолчанию, файл PDBx / mmCif)
- «Pdb» (формат PDB)
- «Xml» (формат PMDML / XML)
- «Ммтф» (сильно сжатый)
- «Bundle» (архив в формате PDB для большой структуры)
Чтобы загрузить файл cif, используйте Bio.MMCIF.MMCIFParser, как указано ниже —
>>> parser = MMCIFParser(QUIET = True)
>>> data = parser.get_structure("2FAT", "2FAT.cif")
Здесь, QUIET подавляет предупреждение при разборе файла. get_structure проанализирует файл и вернет структуру с идентификатором 2FAT (первый аргумент).
После выполнения вышеуказанной команды он анализирует файл и печатает возможное предупреждение, если оно доступно.
Теперь проверьте структуру, используя приведенную ниже команду —
>>> data <Structure id = 2FAT> To get the type, use type method as specified below, >>> print(type(data)) <class 'Bio.PDB.Structure.Structure'>
Мы успешно проанализировали файл и получили структуру белка. Мы узнаем подробности о структуре белка и о том, как его получить, в следующей главе.
PDB Parser
Давайте загрузим пример базы данных в формате PDB с сервера pdb, используя следующую команду:
>>> pdbl = PDBList()
>>> pdbl.retrieve_pdb_file('2FAT', pdir = '.', file_format = 'pdb')
Это загрузит указанный файл (pdb2fat.ent) с сервера и сохранит его в текущем рабочем каталоге.
Чтобы загрузить файл pdb, используйте Bio.PDB.PDBParser, как указано ниже —
>>> parser = PDBParser(PERMISSIVE = True, QUIET = True)
>>> data = parser.get_structure("2fat","pdb2fat.ent")
Здесь get_structure похожа на MMCIFParser. Параметр РАЗРЕШЕНИЕ попытается проанализировать данные белка как можно более гибкими.
Теперь проверьте структуру и ее тип с помощью фрагмента кода, приведенного ниже —
>>> data <Structure id = 2fat> >>> print(type(data)) <class 'Bio.PDB.Structure.Structure'>
Хорошо, структура заголовка хранит информацию словаря. Для этого введите следующую команду —
>>> print(data.header.keys()) dict_keys([ 'name', 'head', 'deposition_date', 'release_date', 'structure_method', 'resolution', 'structure_reference', 'journal_reference', 'author', 'compound', 'source', 'keywords', 'journal']) >>>
Чтобы получить имя, используйте следующий код —
>>> print(data.header["name"]) an anti-urokinase plasminogen activator receptor (upar) antibody: crystal structure and binding epitope >>>
Вы также можете проверить дату и разрешение с помощью приведенного ниже кода —
>>> print(data.header["release_date"]) 2006-11-14 >>> print(data.header["resolution"]) 1.77
Структура PDB
Структура PDB состоит из одной модели, содержащей две цепочки.
- цепь L, содержащая количество остатков
- цепь H, содержащая количество остатков
Каждый остаток состоит из нескольких атомов, каждый из которых имеет трехмерную позицию, представленную координатами (x, y, z).
Давайте узнаем, как получить структуру атома подробно в следующем разделе —
модель
Метод Structure.get_models () возвращает итератор для моделей. Это определено ниже —
>>> model = data.get_models() >>> model <generator object get_models at 0x103fa1c80> >>> models = list(model) >>> models [<Model id = 0>] >>> type(models[0]) <class 'Bio.PDB.Model.Model'>
Здесь Модель описывает ровно одну трехмерную конформацию. Он содержит одну или несколько цепочек.
цепь
Метод Model.get_chain () возвращает итератор по цепочкам. Это определено ниже —
>>> chains = list(models[0].get_chains()) >>> chains [<Chain id = L>, <Chain id = H>] >>> type(chains[0]) <class 'Bio.PDB.Chain.Chain'>
Здесь Chain описывает правильную полипептидную структуру, то есть последовательную последовательность связанных остатков.
остаток
Метод Chain.get_residues () возвращает итератор для остатков. Это определено ниже —
>>> residue = list(chains[0].get_residues()) >>> len(residue) 293 >>> residue1 = list(chains[1].get_residues()) >>> len(residue1) 311
Ну, остаток содержит атомы, которые принадлежат аминокислоте.
атомы
Residue.get_atom () возвращает итератор для атомов, как определено ниже —
>>> atoms = list(residue[0].get_atoms()) >>> atoms [<Atom N>, <Atom CA>, <Atom C>, <Atom Ov, <Atom CB>, <Atom CG>, <Atom OD1>, <Atom OD2>]
Атом содержит трехмерную координату атома и называется вектором. Определяется ниже
>>> atoms[0].get_vector() <Vector 18.49, 73.26, 44.16>
Он представляет значения координат x, y и z.
Биопайтон — Мотив Объекты
Мотив последовательности представляет собой последовательность нуклеотидной или аминокислотной последовательности. Мотивы последовательностей образованы трехмерным расположением аминокислот, которые могут быть не смежными. Biopython предоставляет отдельный модуль Bio.motifs для доступа к функциям последовательности мотивов, как указано ниже —
from Bio import motifs
Создание простого мотива ДНК
Давайте создадим простую последовательность мотивов ДНК, используя следующую команду:
>>> from Bio import motifs
>>> from Bio.Seq import Seq
>>> DNA_motif = [ Seq("AGCT"),
... Seq("TCGA"),
... Seq("AACT"),
... ]
>>> seq = motifs.create(DNA_motif)
>>> print(seq) AGCT TCGA AACT
Чтобы посчитать значения последовательности, используйте следующую команду —
>>> print(seq.counts)
0 1 2 3
A: 2.00 1.00 0.00 1.00
C: 0.00 1.00 2.00 0.00
G: 0.00 1.00 1.00 0.00
T: 1.00 0.00 0.00 2.00
Используйте следующий код для подсчета «A» в последовательности:
>>> seq.counts["A", :] (2, 1, 0, 1)
Если вы хотите получить доступ к столбцам счетчиков, используйте команду ниже —
>>> seq.counts[:, 3]
{'A': 1, 'C': 0, 'T': 2, 'G': 0}
Создание логотипа последовательности
Теперь мы обсудим, как создать логотип последовательности.
Рассмотрим следующую последовательность:
AGCTTACG ATCGTACC TTCCGAAT GGTACGTA AAGCTTGG
Вы можете создать свой собственный логотип, используя следующую ссылку — http://weblogo.berkeley.edu/
Добавьте приведенную выше последовательность, создайте новый логотип и сохраните изображение с именем seq.png в папке вашего биопиона.
seq.png
После создания изображения теперь выполните следующую команду —
>>> seq.weblogo("seq.png")
Этот мотив последовательности ДНК представлен как логотип последовательности для LexA-связывающего мотива.
База данных JASPAR
JASPAR — одна из самых популярных баз данных. Он предоставляет средства любого из форматов мотивов для чтения, записи и сканирования последовательностей. Он хранит метаинформацию для каждого мотива. Модуль Bio.motifs содержит специализированный класс jaspar.Motif для представления атрибутов метаинформации .
У этого есть следующие известные типы атрибутов —
- matrix_id — уникальный идентификатор мотива JASPAR
- name — название мотива
- tf_family — семейство мотивов, например ‘Helix-Loop-Helix’
- data_type — тип данных, используемых в мотиве.
Давайте создадим формат сайтов JASPAR, названный в sample.sites в папке biopython. Это определено ниже —
sample.sites >MA0001 ARNT 1 AACGTGatgtccta >MA0001 ARNT 2 CAGGTGggatgtac >MA0001 ARNT 3 TACGTAgctcatgc >MA0001 ARNT 4 AACGTGacagcgct >MA0001 ARNT 5 CACGTGcacgtcgt >MA0001 ARNT 6 cggcctCGCGTGc
В приведенном выше файле мы создали экземпляры мотива. Теперь давайте создадим объект-мотив из приведенных выше примеров —
>>> from Bio import motifs
>>> with open("sample.sites") as handle:
... data = motifs.read(handle,"sites")
...
>>> print(data)
TF name None
Matrix ID None
Matrix:
0 1 2 3 4 5
A: 2.00 5.00 0.00 0.00 0.00 1.00
C: 3.00 0.00 5.00 0.00 0.00 0.00
G: 0.00 1.00 1.00 6.00 0.00 5.00
T: 1.00 0.00 0.00 0.00 6.00 0.00
Здесь данные читают все экземпляры мотива из файла sample.sites.
Чтобы распечатать все экземпляры из данных, используйте команду ниже —
>>> for instance in data.instances: ... print(instance) ... AACGTG CAGGTG TACGTA AACGTG CACGTG CGCGTG
Используйте приведенную ниже команду для подсчета всех значений —
>>> print(data.counts)
0 1 2 3 4 5
A: 2.00 5.00 0.00 0.00 0.00 1.00
C: 3.00 0.00 5.00 0.00 0.00 0.00
G: 0.00 1.00 1.00 6.00 0.00 5.00
T: 1.00 0.00 0.00 0.00 6.00 0.00
>>>
Biopython — модуль BioSQL
BioSQL — это общая схема базы данных, предназначенная главным образом для хранения последовательностей и связанных с ними данных для всего ядра СУБД. Он спроектирован таким образом, что в нем хранятся данные из всех популярных баз данных по биоинформатике, таких как GenBank, Swissport и т. Д. Он также может использоваться для хранения собственных данных.
В настоящее время BioSQL предоставляет специальную схему для следующих баз данных:
- MySQL (biosqldb-mysql.sql)
- PostgreSQL (biosqldb-pg.sql)
- Oracle (biosqldb-ora / *. Sql)
- SQLite (biosqldb-sqlite.sql)
Он также обеспечивает минимальную поддержку баз данных HSQLDB и Derby на основе Java.
BioPython предоставляет очень простые, легкие и расширенные возможности ORM для работы с базой данных на основе BioSQL. BioPython предоставляет модуль BioSQL для выполнения следующих функций:
- Создать / удалить базу данных BioSQL
- Подключиться к базе данных BioSQL
- Проанализируйте базу данных последовательностей, такую как GenBank, Swisport, результат BLAST, результат Entrez и т. Д., И непосредственно загрузите ее в базу данных BioSQL.
- Получить данные последовательности из базы данных BioSQL
- Получить данные таксономии из NCBI BLAST и сохранить их в базе данных BioSQL
- Выполнить любой SQL-запрос к базе данных BioSQL
Обзор схемы базы данных BioSQL
Прежде чем углубляться в BioSQL, давайте разберемся с основами схемы BioSQL. Схема BioSQL предоставляет более 25 таблиц для хранения данных последовательности, функции последовательности, категории последовательности / онтологии и информации о таксономии. Вот некоторые из важных таблиц:
- biodatabase
- bioentry
- biosequence
- seqfeature
- таксон
- taxon_name
- Антология
- срок
- dxref
Создание базы данных BioSQL
В этом разделе мы создадим образец базы данных BioSQL, biosql, используя схему, предоставленную командой BioSQL. Мы будем работать с базой данных SQLite, так как это действительно легко начать и не имеет сложной настройки.
Здесь мы создадим базу данных BioSQL на основе SQLite, используя следующие шаги.
Шаг 1 — Загрузите движок базы данных SQLite и установите его.
Шаг 2 — Загрузите проект BioSQL с URL GitHub. https://github.com/biosql/biosql
Шаг 3 — Откройте консоль и создайте каталог с помощью mkdir и войдите в него.
cd /path/to/your/biopython/sample mkdir sqlite-biosql cd sqlite-biosql
Шаг 4 — Запустите приведенную ниже команду, чтобы создать новую базу данных SQLite.
> sqlite3.exe mybiosql.db SQLite version 3.25.2 2018-09-25 19:08:10 Enter ".help" for usage hints. sqlite>
Шаг 5 — Скопируйте файл biosqldb-sqlite.sql из проекта BioSQL (/ sql / biosqldb-sqlite.sql`) и сохраните его в текущем каталоге.
Шаг 6 — Запустите приведенную ниже команду, чтобы создать все таблицы.
sqlite> .read biosqldb-sqlite.sql
Теперь все таблицы созданы в нашей новой базе данных.
Шаг 7 — Запустите команду ниже, чтобы увидеть все новые таблицы в нашей базе данных.
sqlite> .headers on sqlite> .mode column sqlite> .separator ROW "\n" sqlite> SELECT name FROM sqlite_master WHERE type = 'table'; biodatabase taxon taxon_name ontology term term_synonym term_dbxref term_relationship term_relationship_term term_path bioentry bioentry_relationship bioentry_path biosequence dbxref dbxref_qualifier_value bioentry_dbxref reference bioentry_reference comment bioentry_qualifier_value seqfeature seqfeature_relationship seqfeature_path seqfeature_qualifier_value seqfeature_dbxref location location_qualifier_value sqlite>
Первые три команды являются командами конфигурации для настройки SQLite для отображения результата в отформатированном виде.
Шаг 8 — Скопируйте образец файла GenBank, ls_orchid.gbk, предоставленный командой BioPython https://raw.githubusercontent.com/biopython/biopython/master/Doc/examples/ls_orchid.gbk, в текущий каталог и сохраните его как orchid.gbk ,
Шаг 9 — Создайте скрипт python load_orchid.py, используя приведенный ниже код, и выполните его.
from Bio import SeqIO
from BioSQL import BioSeqDatabase
import os
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
db = server.new_database("orchid")
count = db.load(SeqIO.parse("orchid.gbk", "gb"), True) server.commit()
server.close()
Приведенный выше код анализирует запись в файле и преобразует ее в объекты Python и вставляет ее в базу данных BioSQL. Мы проанализируем код в следующем разделе.
Наконец, мы создали новую базу данных BioSQL и загрузили в нее несколько примеров данных. Мы обсудим важные таблицы в следующей главе.
Простая схема ER
Таблица базы данных находится в верхней части иерархии, и ее основное назначение — организовать набор данных последовательности в одну группу / виртуальную базу данных. Каждая запись в базе данных biod относится к отдельной базе данных и не смешивается с другой базой данных. Все связанные таблицы в базе данных BioSQL имеют ссылки на запись в базе данных.
Таблица bioentry содержит все детали о последовательности, кроме данных о последовательности. Данные о последовательности конкретной био-записи будут сохранены в таблице биопоследовательности .
taxon и taxon_name являются деталями таксономии, и каждая запись ссылается на эту таблицу для указания информации о таксонах.
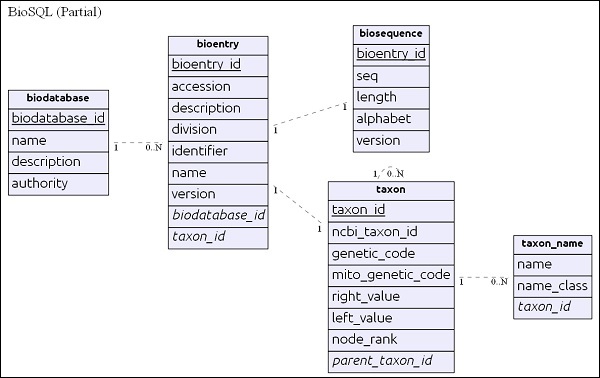
После понимания схемы, давайте посмотрим на некоторые запросы в следующем разделе.
Запросы BioSQL
Давайте углубимся в некоторые SQL-запросы, чтобы лучше понять, как организованы данные и как таблицы связаны друг с другом. Прежде чем продолжить, давайте откроем базу данных с помощью приведенной ниже команды и установим некоторые команды форматирования:
> sqlite3 orchid.db SQLite version 3.25.2 2018-09-25 19:08:10 Enter ".help" for usage hints. sqlite> .header on sqlite> .mode columns
.header и .mode — параметры форматирования для лучшей визуализации данных . Вы также можете использовать любой редактор SQLite для запуска запроса.
Перечислите базу данных виртуальных последовательностей, доступную в системе, как указано ниже —
select * from biodatabase; *** Result *** sqlite> .width 15 15 15 15 sqlite> select * from biodatabase; biodatabase_id name authority description --------------- --------------- --------------- --------------- 1 orchid sqlite>
Здесь у нас есть только одна база данных, орхидея .
Перечислите записи (топ 3), доступные в базе данных orchid с приведенным ниже кодом
select
be.*,
bd.name
from
bioentry be
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid' Limit 1,
3;
*** Result ***
sqlite> .width 15 15 10 10 10 10 10 50 10 10
sqlite> select be.*, bd.name from bioentry be inner join biodatabase bd on
bd.biodatabase_id = be.biodatabase_id where bd.name = 'orchid' Limit 1,3;
bioentry_id biodatabase_id taxon_id name accession identifier division description version name
--------------- --------------- ---------- ---------- ---------- ---------- ----------
---------- ---------- ----------- ---------- --------- ---------- ----------
2 1 19 Z78532 Z78532 2765657 PLN
C.californicum 5.8S rRNA gene and ITS1 and ITS2 DN 1
orchid
3 1 20 Z78531 Z78531 2765656 PLN
C.fasciculatum 5.8S rRNA gene and ITS1 and ITS2 DN 1
orchid
4 1 21 Z78530 Z78530 2765655 PLN
C.margaritaceum 5.8S rRNA gene and ITS1 and ITS2 D 1
orchid
sqlite>
Перечислите подробности последовательности, связанные с записью (номер доступа — Z78530, название — ген C. Fasciculatum 5.8S рРНК и ДНК ITS1 и ITS2) с указанным кодом —
select
substr(cast(bs.seq as varchar), 0, 10) || '...' as seq,
bs.length,
be.accession,
be.description,
bd.name
from
biosequence bs
inner join
bioentry be
on be.bioentry_id = bs.bioentry_id
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid'
and be.accession = 'Z78532';
*** Result ***
sqlite> .width 15 5 10 50 10
sqlite> select substr(cast(bs.seq as varchar), 0, 10) || '...' as seq,
bs.length, be.accession, be.description, bd.name from biosequence bs inner
join bioentry be on be.bioentry_id = bs.bioentry_id inner join biodatabase bd
on bd.biodatabase_id = be.biodatabase_id where bd.name = 'orchid' and
be.accession = 'Z78532';
seq length accession description name
------------ ---------- ---------- ------------ ------------ ---------- ---------- -----------------
CGTAACAAG... 753 Z78532 C.californicum 5.8S rRNA gene and ITS1 and ITS2 DNA orchid
sqlite>
Получите полную последовательность, связанную с записью (инвентарный номер — Z78530, название — ген C. Fasciculatum 5.8S рРНК и ДНК ITS1 и ITS2), используя приведенный ниже код —
select
bs.seq
from
biosequence bs
inner join
bioentry be
on be.bioentry_id = bs.bioentry_id
inner join
biodatabase bd
on bd.biodatabase_id = be.biodatabase_id
where
bd.name = 'orchid'
and be.accession = 'Z78532';
*** Result ***
sqlite> .width 1000
sqlite> select bs.seq from biosequence bs inner join bioentry be on
be.bioentry_id = bs.bioentry_id inner join biodatabase bd on bd.biodatabase_id =
be.biodatabase_id where bd.name = 'orchid' and be.accession = 'Z78532';
seq
----------------------------------------------------------------------------------------
----------------------------
CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGTTGAGACAACAGAATATATGATCGAGTGAATCT
GGAGGACCTGTGGTAACTCAGCTCGTCGTGGCACTGCTTTTGTCGTGACCCTGCTTTGTTGTTGGGCCTCC
TCAAGAGCTTTCATGGCAGGTTTGAACTTTAGTACGGTGCAGTTTGCGCCAAGTCATATAAAGCATCACTGATGAATGACATTATTGT
CAGAAAAAATCAGAGGGGCAGTATGCTACTGAGCATGCCAGTGAATTTTTATGACTCTCGCAACGGATATCTTGGCTC
TAACATCGATGAAGAACGCAG
sqlite>
Список таксонов, связанных с био базой орхидей
select distinct
tn.name
from
biodatabase d
inner join
bioentry e
on e.biodatabase_id = d.biodatabase_id
inner join
taxon t
on t.taxon_id = e.taxon_id
inner join
taxon_name tn
on tn.taxon_id = t.taxon_id
where
d.name = 'orchid' limit 10;
*** Result ***
sqlite> select distinct tn.name from biodatabase d inner join bioentry e on
e.biodatabase_id = d.biodatabase_id inner join taxon t on t.taxon_id =
e.taxon_id inner join taxon_name tn on tn.taxon_id = t.taxon_id where d.name =
'orchid' limit 10;
name
------------------------------
Cypripedium irapeanum
Cypripedium californicum
Cypripedium fasciculatum
Cypripedium margaritaceum
Cypripedium lichiangense
Cypripedium yatabeanum
Cypripedium guttatum
Cypripedium acaule
pink lady's slipper
Cypripedium formosanum
sqlite>
Загрузка данных в базу данных BioSQL
Давайте узнаем, как загрузить данные последовательности в базу данных BioSQL в этой главе. У нас уже есть код для загрузки данных в базу данных в предыдущем разделе, и код выглядит следующим образом:
from Bio import SeqIO
from BioSQL import BioSeqDatabase
import os
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
DBSCHEMA = "biosqldb-sqlite.sql"
SQL_FILE = os.path.join(os.getcwd(), DBSCHEMA)
server.load_database_sql(SQL_FILE)
server.commit()
db = server.new_database("orchid")
count = db.load(SeqIO.parse("orchid.gbk", "gb"), True) server.commit()
server.close()
Мы будем более подробно рассматривать каждую строку кода и его цель —
Линия 1 — загружает модуль SeqIO.
Строка 2 — загружает модуль BioSeqDatabase. Этот модуль предоставляет все функциональные возможности для взаимодействия с базой данных BioSQL.
Линия 3 — Загрузка модуля ОС.
Строка 5 — open_database открывает указанную базу данных (db) с настроенным драйвером (драйвер) и возвращает дескриптор базы данных BioSQL (сервер). Biopython поддерживает базы данных sqlite, mysql, postgresql и oracle.
Строка 6-10 — метод load_database_sql загружает sql из внешнего файла и выполняет его. Метод commit фиксирует транзакцию. Мы можем пропустить этот шаг, потому что мы уже создали базу данных со схемой.
Строка 12 — методы new_database создают новую виртуальную базу данных orchid и возвращают дескриптор db для выполнения команды в базе данных orchid.
Строка 13 — метод load загружает записи последовательности (итерируемый SeqRecord) в базу данных orchid. SqlIO.parse анализирует базу данных GenBank и возвращает все последовательности в ней как повторяемый SeqRecord. Второй параметр (True) в методе загрузки указывает ему на получение сведений о таксономии данных последовательности с веб-сайта взрыва NCBI, если они еще не доступны в системе.
Строка 14 — commit фиксирует транзакцию.
Строка 15 — close закрывает соединение с базой данных и уничтожает дескриптор сервера.
Получить данные последовательности
Давайте выберем последовательность с идентификатором 2765658 из базы данных орхидей, как показано ниже:
from BioSQL import BioSeqDatabase
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
db = server["orchid"]
seq_record = db.lookup(gi = 2765658)
print(seq_record.id, seq_record.description[:50] + "...")
print("Sequence length %i," % len(seq_record.seq))
Здесь server [«orchid»] возвращает дескриптор для извлечения данных из виртуальной базы данных orchid. Метод lookup предоставляет возможность выбирать последовательности на основе критериев, и мы выбрали последовательность с идентификатором 2765658. lookup возвращает информацию о последовательности в виде SeqRecordobject. Поскольку мы уже знаем, как работать с SeqRecord`, легко получить данные из него.
Удалить базу данных
Удалить базу данных так же просто, как вызвать метод remove_database с правильным именем базы данных, а затем зафиксировать его, как указано ниже —
from BioSQL import BioSeqDatabase
server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db")
server.remove_database("orchids")
server.commit()
Биопайтон — популяционная генетика
Генетика населения играет важную роль в теории эволюции. Он анализирует генетические различия между видами, а также двух или более особей в пределах одного и того же вида.
Biopython предоставляет модуль Bio.PopGen для популяционной генетики и в основном поддерживает `GenePop, популярный генетический пакет, разработанный Мишелем Рэймондом и Франсуа Руссе.
Простой парсер
Давайте напишем простое приложение для разбора формата GenePop и понимания концепции.
Загрузите файл genePop, предоставленный командой Biopython, по ссылке, приведенной ниже — https://raw.githubusercontent.com/biopython/biopython/master/Tests/PopGen/c3line.gen.
Загрузите модуль GenePop, используя приведенный ниже фрагмент кода —
from Bio.PopGen import GenePop
Разобрать файл, используя метод GenePop.read, как показано ниже —
record = GenePop.read(open("c3line.gen"))
Показать информацию о местах и населении, как указано ниже —
>>> record.loci_list
['136255903', '136257048', '136257636']
>>> record.pop_list
['4', 'b3', '5']
>>> record.populations
[[('1', [(3, 3), (4, 4), (2, 2)]), ('2', [(3, 3), (3, 4), (2, 2)]),
('3', [(3, 3), (4, 4), (2, 2)]), ('4', [(3, 3), (4, 3), (None, None)])],
[('b1', [(None, None), (4, 4), (2, 2)]), ('b2', [(None, None), (4, 4), (2, 2)]),
('b3', [(None, None), (4, 4), (2, 2)])],
[('1', [(3, 3), (4, 4), (2, 2)]), ('2', [(3, 3), (1, 4), (2, 2)]),
('3', [(3, 2), (1, 1), (2, 2)]), ('4',
[(None, None), (4, 4), (2, 2)]), ('5', [(3, 3), (4, 4), (2, 2)])]]
>>>
Здесь в файле доступны три локуса и три группы населения: первая популяция имеет 4 записи, вторая популяция имеет 3 записи и третья популяция имеет 5 записей. record.populations показывает все группы населения с данными по аллелям для каждого локуса.
Манипулировать файлом GenePop
Biopython предоставляет опции для удаления данных о местонахождении и населении.
Удалить население, установленное положением,
>>> record.remove_population(0)
>>> record.populations
[[('b1', [(None, None), (4, 4), (2, 2)]),
('b2', [(None, None), (4, 4), (2, 2)]),
('b3', [(None, None), (4, 4), (2, 2)])],
[('1', [(3, 3), (4, 4), (2, 2)]),
('2', [(3, 3), (1, 4), (2, 2)]),
('3', [(3, 2), (1, 1), (2, 2)]),
('4', [(None, None), (4, 4), (2, 2)]),
('5', [(3, 3), (4, 4), (2, 2)])]]
>>>
Удалить локус по положению,
>>> record.remove_locus_by_position(0)
>>> record.loci_list
['136257048', '136257636']
>>> record.populations
[[('b1', [(4, 4), (2, 2)]), ('b2', [(4, 4), (2, 2)]), ('b3', [(4, 4), (2, 2)])],
[('1', [(4, 4), (2, 2)]), ('2', [(1, 4), (2, 2)]),
('3', [(1, 1), (2, 2)]), ('4', [(4, 4), (2, 2)]), ('5', [(4, 4), (2, 2)])]]
>>>
Удалить локус по имени,
>>> record.remove_locus_by_name('136257636') >>> record.loci_list
['136257048']
>>> record.populations
[[('b1', [(4, 4)]), ('b2', [(4, 4)]), ('b3', [(4, 4)])],
[('1', [(4, 4)]), ('2', [(1, 4)]),
('3', [(1, 1)]), ('4', [(4, 4)]), ('5', [(4, 4)])]]
>>>
Интерфейс с программным обеспечением GenePop
Biopython предоставляет интерфейсы для взаимодействия с программным обеспечением GenePop и тем самым предоставляет множество функциональных возможностей. Для этого используется модуль Bio.PopGen.GenePop. Одним из таких простых в использовании интерфейс является EasyController. Давайте проверим, как анализировать файл GenePop и провести анализ с помощью EasyController.
Сначала установите программное обеспечение GenePop и поместите папку установки в системный путь. Чтобы получить основную информацию о файле GenePop, создайте объект EasyController и затем вызовите метод get_basic_info, как указано ниже —
>>> from Bio.PopGen.GenePop.EasyController import EasyController
>>> ec = EasyController('c3line.gen')
>>> print(ec.get_basic_info())
(['4', 'b3', '5'], ['136255903', '136257048', '136257636'])
>>>
Здесь первый элемент — список населения, а второй элемент — список локусов.
Чтобы получить весь список аллелей определенного локуса, вызовите метод get_alleles_all_pops, передав имя локуса, как указано ниже —
>>> allele_list = ec.get_alleles_all_pops("136255903")
>>> print(allele_list)
[2, 3]
Чтобы получить список аллелей по определенной популяции и локусу, вызовите get_alleles, передавая имя локуса и положение популяции, как указано ниже —
>>> allele_list = ec.get_alleles(0, "136255903") >>> print(allele_list) [] >>> allele_list = ec.get_alleles(1, "136255903") >>> print(allele_list) [] >>> allele_list = ec.get_alleles(2, "136255903") >>> print(allele_list) [2, 3] >>>
Аналогичным образом EasyController предоставляет множество функций: частоту аллелей, частоту генотипа, статистику мультилокусных F, равновесие Харди-Вайнберга, неравновесие по сцеплению и т. Д.
Биопайтон — Анализ генома
Геном — это полный набор ДНК, включая все его гены. Анализ генома относится к изучению отдельных генов и их роли в наследовании.
Геном Диаграмма
Геномная диаграмма представляет генетическую информацию в виде диаграмм. Biopython использует модуль Bio.Graphics.GenomeDiagram для представления GenomeDiagram. Модуль GenomeDiagram требует установки ReportLab.
Шаги для создания диаграммы
Процесс создания диаграммы обычно следует следующему простому шаблону —
-
Создайте FeatureSet для каждого отдельного набора объектов, которые вы хотите отобразить, и добавьте к ним объекты Bio.SeqFeature.
-
Создайте GraphSet для каждого графика, который вы хотите отобразить, и добавьте к ним данные графика.
-
Создайте дорожку для каждой дорожки, которую вы хотите на диаграмме, и добавьте GraphSets и FeatureSets к нужным дорожкам.
-
Создайте диаграмму и добавьте к ней дорожки.
-
Скажите Диаграмму, чтобы нарисовать изображение.
-
Запишите изображение в файл.
Создайте FeatureSet для каждого отдельного набора объектов, которые вы хотите отобразить, и добавьте к ним объекты Bio.SeqFeature.
Создайте GraphSet для каждого графика, который вы хотите отобразить, и добавьте к ним данные графика.
Создайте дорожку для каждой дорожки, которую вы хотите на диаграмме, и добавьте GraphSets и FeatureSets к нужным дорожкам.
Создайте диаграмму и добавьте к ней дорожки.
Скажите Диаграмму, чтобы нарисовать изображение.
Запишите изображение в файл.
Давайте возьмем пример входного файла GenBank —
https://raw.githubusercontent.com/biopython/biopython/master/Doc/examples/ls_orchid.gbk и прочитайте записи из объекта SeqRecord, а затем, наконец, нарисуйте диаграмму генома. Это объясняется ниже,
Сначала мы импортируем все модули, как показано ниже —
>>> from reportlab.lib import colors >>> from reportlab.lib.units import cm >>> from Bio.Graphics import GenomeDiagram
Теперь импортируйте модуль SeqIO для чтения данных —
>>> from Bio import SeqIO
record = SeqIO.read("example.gb", "genbank")
Здесь запись читает последовательность из файла genbank.
Теперь создайте пустую диаграмму, чтобы добавить трек и набор функций —
>>> diagram = GenomeDiagram.Diagram( "Yersinia pestis biovar Microtus plasmid pPCP1") >>> track = diagram.new_track(1, name="Annotated Features") >>> feature = track.new_set()
Теперь мы можем применить изменения цветовой темы, используя альтернативные цвета от зеленого до серого, как определено ниже —
>>> for feature in record.features: >>> if feature.type != "gene": >>> continue >>> if len(feature) % 2 == 0: >>> color = colors.blue >>> else: >>> color = colors.red >>> >>> feature.add_feature(feature, color=color, label=True)
Теперь вы можете увидеть ответ ниже на вашем экране —
<Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d3dc90> <Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d3dfd0> <Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x1007627d0> <Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57290> <Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57050> <Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57390> <Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57590> <Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57410> <Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d57490> <Bio.Graphics.GenomeDiagram._Feature.Feature object at 0x105d574d0>
Давайте нарисуем диаграмму для вышеуказанных входных записей —
>>> diagram.draw(
format = "linear", orientation = "landscape", pagesize = 'A4',
... fragments = 4, start = 0, end = len(record))
>>> diagram.write("orchid.pdf", "PDF")
>>> diagram.write("orchid.eps", "EPS")
>>> diagram.write("orchid.svg", "SVG")
>>> diagram.write("orchid.png", "PNG")
После выполнения вышеуказанной команды вы можете увидеть следующее изображение, сохраненное в вашем каталоге Biopython.
** Result ** genome.png
Вы также можете нарисовать изображение в круговом формате, внеся следующие изменения:
>>> diagram.draw(
format = "circular", circular = True, pagesize = (20*cm,20*cm),
... start = 0, end = len(record), circle_core = 0.7)
>>> diagram.write("circular.pdf", "PDF")
Обзор хромосом
Молекула ДНК упакована в нитевидные структуры, называемые хромосомами. Каждая хромосома состоит из ДНК, плотно обмотанной много раз вокруг белков, называемых гистонами, которые поддерживают ее структуру.
Хромосомы не видны в ядре клетки — даже под микроскопом — когда клетка не делится. Тем не менее, ДНК, которая составляет хромосомы, становится более плотно упакованной во время клеточного деления и затем видна под микроскопом.
У людей каждая клетка обычно содержит 23 пары хромосом, что в общей сложности составляет 46. Двадцать две из этих пар, называемые аутосомами, выглядят одинаково как у мужчин, так и у женщин. 23-я пара, половые хромосомы, различается между мужчинами и женщинами. У самок есть две копии Х-хромосомы, а у самцов — одна Х и одна Y-хромосома.
Биопайтон — фенотип микрочипа
Фенотип определяется как наблюдаемый характер или признак, проявляемый организмом в отношении определенного химического вещества или окружающей среды. Фенотип микрочипа одновременно измеряет реакцию организма на большее количество химических веществ и окружающей среды и анализирует данные, чтобы понять генную мутацию, генные признаки и т. Д.
Biopython предоставляет отличный модуль Bio.Phenotype для анализа фенотипических данных. Давайте узнаем, как анализировать, интерполировать, извлекать и анализировать данные микрочипов фенотипа в этой главе.
анализ
Данные микрочипа фенотипа могут быть в двух форматах: CSV и JSON. Biopython поддерживает оба формата. Анализатор Biopython анализирует данные микрочипа фенотипа и возвращает их в виде коллекции объектов PlateRecord. Каждый объект PlateRecord содержит коллекцию объектов WellRecord. Каждый объект WellRecord содержит данные в формате 8 строк и 12 столбцов. Восемь строк представлены от A до H, а 12 столбцов представлены от 01 до 12. Например, 4- й ряд и 6- й столбец представлены D06.
Давайте разберемся с форматом и концепцией разбора на следующем примере:
Шаг 1 — Загрузите файл Plates.csv, предоставленный командой Biopython — https://raw.githubusercontent.com/biopython/biopython/master/Doc/examples/Plates.csv
Шаг 2 — Загрузите модуль фенотипа, как показано ниже —
>>> from Bio import phenotype
Шаг 3 — Вызвать метод phenotype.parse, передав файл данных и опцию формата («pm-csv»). Он возвращает итеративный PlateRecord, как показано ниже,
>>> plates = list(phenotype.parse('Plates.csv', "pm-csv"))
>>> plates
[PlateRecord('WellRecord['A01'], WellRecord['A02'], WellRecord['A03'], ..., WellRecord['H12']'),
PlateRecord('WellRecord['A01'], WellRecord['A02'], WellRecord['A03'], ..., WellRecord['H12']'),
PlateRecord('WellRecord['A01'], WellRecord['A02'], WellRecord['A03'], ..., WellRecord['H12']'),
PlateRecord('WellRecord['A01'], WellRecord['A02'],WellRecord['A03'], ..., WellRecord['H12']')]
>>>
Шаг 4 — Доступ к первой табличке из списка, как показано ниже —
>>> plate = plates[0]
>>> plate
PlateRecord('WellRecord['A01'], WellRecord['A02'], WellRecord['A03'], ...,
WellRecord['H12']')
>>>
Шаг 5 — Как обсуждалось ранее, пластина содержит 8 рядов, каждый из которых содержит 12 предметов. Доступ к WellRecord можно получить двумя способами, как указано ниже:
>>> well = plate["A04"]
>>> well = plate[0, 4]
>>> well WellRecord('(0.0, 0.0), (0.25, 0.0), (0.5, 0.0), (0.75, 0.0),
(1.0, 0.0), ..., (71.75, 388.0)')
>>>
Шаг 6 — Каждая скважина будет иметь серии измерений в разные моменты времени, и к ней можно получить доступ, используя цикл for, как указано ниже —
>>> for v1, v2 in well: ... print(v1, v2) ... 0.0 0.0 0.25 0.0 0.5 0.0 0.75 0.0 1.0 0.0 ... 71.25 388.0 71.5 388.0 71.75 388.0 >>>
интерполирование
Интерполяция дает более глубокое понимание данных. Biopython предоставляет методы для интерполяции данных WellRecord для получения информации о промежуточных временных точках. Синтаксис похож на индексирование списка и поэтому прост в изучении.
Чтобы получить данные за 20,1 часа, просто передайте в качестве значений индекса, как указано ниже —
>>> well[20.10] 69.40000000000003 >>>
Мы можем передать начальный и конечный момент времени, как указано ниже —
>>> well[20:30] [67.0, 84.0, 102.0, 119.0, 135.0, 147.0, 158.0, 168.0, 179.0, 186.0] >>>
Приведенная выше команда интерполирует данные от 20 часов до 30 часов с интервалом в 1 час. По умолчанию интервал составляет 1 час, и мы можем изменить его на любое значение. Например, давайте дадим интервал 15 минут (0,25 часа), как указано ниже —
>>> well[20:21:0.25] [67.0, 73.0, 75.0, 81.0] >>>
Анализировать и извлекать
Biopython предоставляет метод, пригодный для анализа данных WellRecord с использованием сигмоидальных функций Gompertz, Logistic и Richards. По умолчанию метод fit использует функцию Gompertz. Нам нужно вызвать метод fit объекта WellRecord, чтобы выполнить задачу. Кодировка выглядит следующим образом —
>>> well.fit() Traceback (most recent call last): ... Bio.MissingPythonDependencyError: Install scipy to extract curve parameters. >>> well.model >>> getattr(well, 'min') 0.0 >>> getattr(well, 'max') 388.0 >>> getattr(well, 'average_height') 205.42708333333334 >>>
Biopython зависит от модуля scipy, чтобы выполнить расширенный анализ. Он будет вычислять минимальные, максимальные и средние значения высоты без использования модуля scipy.
Биопайтон — черчение
В этой главе рассказывается о том, как строить последовательности. Прежде чем перейти к этой теме, давайте разберемся с основами черчения.
Заговор
Matplotlib — это библиотека графиков Python, которая производит качественные рисунки в различных форматах. Мы можем создавать различные типы графиков, таких как линейные диаграммы, гистограммы, гистограммы, круговые диаграммы, точечные диаграммы и т. Д.
pyLab — это модуль, который принадлежит matplotlib, который объединяет числовой модуль numpy с графическим модулем построения графиков pyplot. Biopython использует модуль pylab для построения последовательностей. Для этого нам нужно импортировать приведенный ниже код —
import pylab
Перед импортом нам нужно установить пакет matplotlib с помощью команды pip с командой, приведенной ниже —
pip install matplotlib
Образец входного файла
Создайте образец файла с именем plot.fasta в вашем каталоге Biopython и добавьте следующие изменения:
>seq0 FQTWEEFSRAAEKLYLADPMKVRVVLKYRHVDGNLCIKVTDDLVCLVYRTDQAQDVKKIEKF >seq1 KYRTWEEFTRAAEKLYQADPMKVRVVLKYRHCDGNLCIKVTDDVVCLLYRTDQAQDVKKIEKFHSQLMRLME >seq2 EEYQTWEEFARAAEKLYLTDPMKVRVVLKYRHCDGNLCMKVTDDAVCLQYKTDQAQDVKKVEKLHGK >seq3 MYQVWEEFSRAVEKLYLTDPMKVRVVLKYRHCDGNLCIKVTDNSVCLQYKTDQAQDV >seq4 EEFSRAVEKLYLTDPMKVRVVLKYRHCDGNLCIKVTDNSVVSYEMRLFGVQKDNFALEHSLL >seq5 SWEEFAKAAEVLYLEDPMKCRMCTKYRHVDHKLVVKLTDNHTVLKYVTDMAQDVKKIEKLTTLLMR >seq6 FTNWEEFAKAAERLHSANPEKCRFVTKYNHTKGELVLKLTDDVVCLQYSTNQLQDVKKLEKLSSTLLRSI >seq7 SWEEFVERSVQLFRGDPNATRYVMKYRHCEGKLVLKVTDDRECLKFKTDQAQDAKKMEKLNNIFF >seq8 SWDEFVDRSVQLFRADPESTRYVMKYRHCDGKLVLKVTDNKECLKFKTDQAQEAKKMEKLNNIFFTLM >seq9 KNWEDFEIAAENMYMANPQNCRYTMKYVHSKGHILLKMSDNVKCVQYRAENMPDLKK >seq10 FDSWDEFVSKSVELFRNHPDTTRYVVKYRHCEGKLVLKVTDNHECLKFKTDQAQDAKKMEK
Линия Участок
Теперь давайте создадим простой линейный график для вышеуказанного файла fasta.
Шаг 1 — Импортируйте модуль SeqIO для чтения файла fasta.
>>> from Bio import SeqIO
Шаг 2 — Разбор входного файла.
>>> records = [len(rec) for rec in SeqIO.parse("plot.fasta", "fasta")]
>>> len(records)
11
>>> max(records)
72
>>> min(records)
57
Шаг 3 — Давайте импортируем модуль pylab.
>>> import pylab
Шаг 4 — Сконфигурируйте линейный график, назначив метки оси x и y.
>>> pylab.xlabel("sequence length")
Text(0.5, 0, 'sequence length')
>>> pylab.ylabel("count")
Text(0, 0.5, 'count')
>>>
Шаг 5 — Настройте линейный график, настроив отображение сетки.
>>> pylab.grid()
Шаг 6 — Нарисуйте простую линейную диаграмму, вызвав метод plot и предоставив записи в качестве входных данных.
>>> pylab.plot(records) [<matplotlib.lines.Line2D object at 0x10b6869d 0>]
Шаг 7 — Наконец, сохраните диаграмму, используя приведенную ниже команду.
>>> pylab.savefig("lines.png")
Результат
После выполнения вышеуказанной команды вы можете увидеть следующее изображение, сохраненное в вашем каталоге Biopython.
Гистограмма
Гистограмма используется для непрерывных данных, где ячейки представляют диапазоны данных. Гистограмма рисования такая же, как линейная диаграмма, за исключением pylab.plot. Вместо этого вызовите метод Hist модуля pylab с записями и некоторым значением custum для bin (5). Полное кодирование выглядит следующим образом:
Шаг 1 — Импортируйте модуль SeqIO для чтения файла fasta.
>>> from Bio import SeqIO
Шаг 2 — Разбор входного файла.
>>> records = [len(rec) for rec in SeqIO.parse("plot.fasta", "fasta")]
>>> len(records)
11
>>> max(records)
72
>>> min(records)
57
Шаг 3 — Давайте импортируем модуль pylab.
>>> import pylab
Шаг 4 — Сконфигурируйте линейный график, назначив метки оси x и y.
>>> pylab.xlabel("sequence length")
Text(0.5, 0, 'sequence length')
>>> pylab.ylabel("count")
Text(0, 0.5, 'count')
>>>
Шаг 5 — Настройте линейный график, настроив отображение сетки.
>>> pylab.grid()
Шаг 6 — Нарисуйте простую линейную диаграмму, вызвав метод plot и предоставив записи в качестве входных данных.
>>> pylab.hist(records,bins=5) (array([2., 3., 1., 3., 2.]), array([57., 60., 63., 66., 69., 72.]), <a list of 5 Patch objects>) >>>
Шаг 7 — Наконец, сохраните диаграмму, используя приведенную ниже команду.
>>> pylab.savefig("hist.png")
Результат
После выполнения вышеуказанной команды вы можете увидеть следующее изображение, сохраненное в вашем каталоге Biopython.
Процент GC в последовательности
Процент GC является одним из наиболее часто используемых аналитических данных для сравнения различных последовательностей. Мы можем сделать простую линейную диаграмму, используя GC Percentage из набора последовательностей, и сразу же сравнить ее. Здесь мы можем просто изменить данные с длины последовательности на процент GC. Полная кодировка приведена ниже —
Шаг 1 — Импортируйте модуль SeqIO для чтения файла fasta.
>>> from Bio import SeqIO
Шаг 2 — Разбор входного файла.
>>> from Bio.SeqUtils import GC
>>> gc = sorted(GC(rec.seq) for rec in SeqIO.parse("plot.fasta", "fasta"))
Шаг 3 — Давайте импортируем модуль pylab.
>>> import pylab
Шаг 4 — Сконфигурируйте линейный график, назначив метки оси x и y.
>>> pylab.xlabel("Genes")
Text(0.5, 0, 'Genes')
>>> pylab.ylabel("GC Percentage")
Text(0, 0.5, 'GC Percentage')
>>>
Шаг 5 — Настройте линейный график, настроив отображение сетки.
>>> pylab.grid()
Шаг 6 — Нарисуйте простую линейную диаграмму, вызвав метод plot и предоставив записи в качестве входных данных.
>>> pylab.plot(gc) [<matplotlib.lines.Line2D object at 0x10b6869d 0>]
Шаг 7 — Наконец, сохраните диаграмму, используя приведенную ниже команду.
>>> pylab.savefig("gc.png")
Результат
После выполнения вышеуказанной команды вы можете увидеть следующее изображение, сохраненное в вашем каталоге Biopython.
Биопайтон — кластерный анализ
Как правило, кластерный анализ группирует набор объектов в одну группу. Эта концепция в основном используется в интеллектуальном анализе данных, статистическом анализе данных, машинном обучении, распознавании образов, анализе изображений, биоинформатике и т. Д. С помощью различных алгоритмов можно понять, как кластер широко используется в различных анализах.
Согласно Биоинформатике, кластерный анализ в основном используется при анализе данных экспрессии генов, чтобы найти группы генов с одинаковой экспрессией генов.
В этой главе мы рассмотрим важные алгоритмы в Biopython, чтобы понять основы кластеризации на реальном наборе данных.
Biopython использует модуль Bio.Cluster для реализации всех алгоритмов. Поддерживаются следующие алгоритмы —
- Иерархическая кластеризация
- K — кластеризация
- Самоорганизующиеся карты
- Анализ главных компонентов
Давайте кратко познакомимся с приведенными выше алгоритмами.
Иерархическая кластеризация
Иерархическая кластеризация используется, чтобы связать каждый узел мерой расстояния с ближайшим соседом и создать кластер. Узел Bio.Cluster имеет три атрибута: левый, правый и расстояние. Давайте создадим простой кластер, как показано ниже —
>>> from Bio.Cluster import Node >>> n = Node(1,10) >>> n.left = 11 >>> n.right = 0 >>> n.distance = 1 >>> print(n) (11, 0): 1
Если вы хотите построить кластеризацию на основе дерева, используйте команду ниже —
>>> n1 = [Node(1, 2, 0.2), Node(0, -1, 0.5)] >>> n1_tree = Tree(n1) >>> print(n1_tree) (1, 2): 0.2 (0, -1): 0.5 >>> print(n1_tree[0]) (1, 2): 0.2
Давайте выполним иерархическую кластеризацию с помощью модуля Bio.Cluster.
Рассмотрим расстояние, определенное в массиве.
>>> import numpy as np >>> distance = array([[1,2,3],[4,5,6],[3,5,7]])
Теперь добавьте массив расстояний в кластер дерева.
>>> from Bio.Cluster import treecluster >>> cluster = treecluster(distance) >>> print(cluster) (2, 1): 0.666667 (-1, 0): 9.66667
Вышеприведенная функция возвращает объект кластера Tree. Этот объект содержит узлы, в которых количество элементов сгруппировано в виде строк или столбцов.
K — кластеризация
Это тип алгоритма разбиения, который классифицируется на k — средние, медианы и кластеры медоидов. Давайте разберемся с каждым из кластеров вкратце.
K-означает кластеризацию
Этот подход популярен в интеллектуальном анализе данных. Цель этого алгоритма — найти группы в данных, количество групп которых представлено переменной K.
Алгоритм работает итеративно, чтобы назначить каждую точку данных одной из K групп на основе предоставленных функций. Точки данных сгруппированы на основе сходства признаков.
>>> from Bio.Cluster import kcluster >>> from numpy import array >>> data = array([[1, 2], [3, 4], [5, 6]]) >>> clusterid, error,found = kcluster(data) >>> print(clusterid) [0 0 1] >>> print(found) 1
К-медианы кластеризации
Это еще один тип алгоритма кластеризации, который вычисляет среднее значение для каждого кластера, чтобы определить его центроид.
Кластеризация K-medoids
Этот подход основан на заданном наборе элементов с использованием матрицы расстояний и количества кластеров, переданных пользователем.
Рассмотрим матрицу расстояний, как определено ниже —
>>> distance = array([[1,2,3],[4,5,6],[3,5,7]])
Мы можем рассчитать кластеризацию k-medoids с помощью следующей команды —
>>> from Bio.Cluster import kmedoids >>> clusterid, error, found = kmedoids(distance)
Давайте рассмотрим пример.
Функция kcluster принимает матрицу данных в качестве входных данных, а не экземпляры Seq. Вам нужно преобразовать свои последовательности в матрицу и передать это функции kcluster.
Одним из способов преобразования данных в матрицу, содержащую только числовые элементы, является использование функции numpy.fromstring . Он в основном переводит каждую букву в последовательности в ее аналог ASCII.
Это создает двумерный массив кодированных последовательностей, который функция kcluster распознает и использует для кластеризации ваших последовательностей.
>>> from Bio.Cluster import kcluster >>> import numpy as np >>> sequence = [ 'AGCT','CGTA','AAGT','TCCG'] >>> matrix = np.asarray([np.fromstring(s, dtype=np.uint8) for s in sequence]) >>> clusterid,error,found = kcluster(matrix) >>> print(clusterid) [1 0 0 1]
Самоорганизующиеся карты
Этот подход представляет собой тип искусственной нейронной сети. Он разработан Кохоненом и часто называется картой Кохонена. Он организует элементы в кластеры на основе прямоугольной топологии.
Давайте создадим простой кластер, используя то же расстояние массива, как показано ниже —
>>> from Bio.Cluster import somcluster >>> from numpy import array >>> data = array([[1, 2], [3, 4], [5, 6]]) >>> clusterid,map = somcluster(data) >>> print(map) [[[-1.36032469 0.38667395]] [[-0.41170578 1.35295911]]] >>> print(clusterid) [[1 0] [1 0] [1 0]]
Здесь clusterid — это массив с двумя столбцами, где количество строк равно количеству кластеризованных элементов, а data — это массив с измерениями, как строками, так и столбцами.
Анализ главных компонентов
Анализ основных компонентов полезен для визуализации многомерных данных. Это метод, который использует простые матричные операции из линейной алгебры и статистики для вычисления проекции исходных данных в то же число или меньшее число измерений.
Анализ главных компонентов возвращает значение столбца кортежа, координаты, компоненты и собственные значения. Давайте посмотрим на основы этой концепции.
>>> from numpy import array >>> from numpy import mean >>> from numpy import cov >>> from numpy.linalg import eig # define a matrix >>> A = array([[1, 2], [3, 4], [5, 6]]) >>> print(A) [[1 2] [3 4] [5 6]] # calculate the mean of each column >>> M = mean(A.T, axis = 1) >>> print(M) [ 3. 4.] # center columns by subtracting column means >>> C = A - M >>> print(C) [[-2. -2.] [ 0. 0.] [ 2. 2.]] # calculate covariance matrix of centered matrix >>> V = cov(C.T) >>> print(V) [[ 4. 4.] [ 4. 4.]] # eigendecomposition of covariance matrix >>> values, vectors = eig(V) >>> print(vectors) [[ 0.70710678 -0.70710678] [ 0.70710678 0.70710678]] >>> print(values) [ 8. 0.]
Давайте применим те же данные прямоугольной матрицы к модулю Bio.Cluster, как определено ниже:
>>> from Bio.Cluster import pca >>> from numpy import array >>> data = array([[1, 2], [3, 4], [5, 6]]) >>> columnmean, coordinates, components, eigenvalues = pca(data) >>> print(columnmean) [ 3. 4.] >>> print(coordinates) [[-2.82842712 0. ] [ 0. 0. ] [ 2.82842712 0. ]] >>> print(components) [[ 0.70710678 0.70710678] [ 0.70710678 -0.70710678]] >>> print(eigenvalues) [ 4. 0.]
Биопайтон — Машинное обучение
Биоинформатика — отличная область для применения алгоритмов машинного обучения. Здесь мы имеем генетическую информацию о большом количестве организмов, и невозможно вручную проанализировать всю эту информацию. Если используется правильный алгоритм машинного обучения, мы можем извлечь много полезной информации из этих данных. Biopython предоставляет полезный набор алгоритмов для контролируемого машинного обучения.
Контролируемое обучение основано на входной переменной (X) и выходной переменной (Y). Он использует алгоритм для изучения функции отображения от входа к выходу. Это определено ниже —
Y = f(X)
Основная цель этого подхода состоит в аппроксимации функции отображения, и когда у вас есть новые входные данные (x), вы можете предсказать выходные переменные (Y) для этих данных.
Модель логистической регрессии
Логистическая регрессия — контролируемый алгоритм машинного обучения. Он используется для определения различий между классами K, используя взвешенную сумму переменных-предикторов. Он вычисляет вероятность возникновения события и может быть использован для обнаружения рака.
Biopython предоставляет модуль Bio.LogisticRegression для прогнозирования переменных на основе алгоритма логистической регрессии. В настоящее время Biopython реализует алгоритм логистической регрессии только для двух классов (K = 2).
k-Ближайшие соседи
k-Ближайшие соседи также являются контролируемым алгоритмом машинного обучения. Он работает путем классификации данных на основе ближайших соседей. Biopython предоставляет модуль Bio.KNN для прогнозирования переменных на основе алгоритма k-ближайших соседей.
Наивный байесовский
Наивные байесовские классификаторы представляют собой набор алгоритмов классификации, основанных на теореме Байеса. Это не отдельный алгоритм, а семейство алгоритмов, в которых все они придерживаются общего принципа, то есть каждая классифицируемая пара признаков не зависит друг от друга. Biopython предоставляет модуль Bio.NaiveBayes для работы с наивным байесовским алгоритмом.
Марковская модель
Марковская модель — это математическая система, определяемая как совокупность случайных величин, в которой происходит переход из одного состояния в другое в соответствии с определенными вероятностными правилами. Biopython предоставляет модули Bio.MarkovModel и Bio.HMM.MarkovModel для работы с марковскими моделями .
Биопайтон — Методы испытаний
Biopython имеет обширный тестовый скрипт для тестирования программного обеспечения в различных условиях, чтобы убедиться, что программное обеспечение не содержит ошибок. Чтобы запустить тестовый скрипт, загрузите исходный код Biopython, а затем выполните следующую команду:
python run_tests.py
Это запустит все тестовые сценарии и даст следующий результат:
Python version: 2.7.12 (v2.7.12:d33e0cf91556, Jun 26 2016, 12:10:39) [GCC 4.2.1 (Apple Inc. build 5666) (dot 3)] Operating system: posix darwin test_Ace ... ok test_Affy ... ok test_AlignIO ... ok test_AlignIO_ClustalIO ... ok test_AlignIO_EmbossIO ... ok test_AlignIO_FastaIO ... ok test_AlignIO_MauveIO ... ok test_AlignIO_PhylipIO ... ok test_AlignIO_convert ... ok ........................................... ...........................................
Мы также можем запустить отдельный тестовый скрипт, как указано ниже —
python test_AlignIO.py
Заключение
Как мы узнали, Biopython является одним из важных программ в области биоинформатики. Будучи написанным на python (легко учиться и писать), он предоставляет широкие функциональные возможности для любых вычислений и операций в области биоинформатики. Он также обеспечивает простой и гибкий интерфейс практически со всем популярным программным обеспечением для биоинформатики, чтобы использовать его функциональность.